V2O5/g⁃C3N4催化剂的制备及其模拟油中硫化物的脱除
2021-04-06李秀萍赵荣祥
张 豪,李秀萍,赵荣祥
(辽宁石油化工大学石油化工学院,辽宁抚顺113001)
随着汽车工业的迅速发展,燃料油燃烧产生的硫化物对环境的污染越来越严重[1⁃2]。因此,油品的深度脱硫受到世界各国的重视。目前,加氢脱硫工艺是油品脱硫的常见方法[3⁃6],但因其操作成本较高、反应条件苛刻,且对噻吩类硫化物脱除效果欠佳,因此非加氢脱硫工艺备受关注。常见的非加氢脱硫工艺包括吸附脱硫、萃取脱硫、氧化脱硫等[7⁃9]。其中,氧化脱硫工艺因具有反应条件温和、对噻吩类脱除效果好的特点而被认为是加氢脱硫的必要补充。
金属氧化物V2O5因具有良好油品脱硫性能而成为研究热点。例如,宋华等[10]以V2O5为催化剂,无水乙醇为萃取剂实现了在温和的条件下对汽油进行氧化脱硫。M.A.Ramos⁃Luna等[11]通过热扩散和湿浸渍法制备了V2O5/TiO2催化剂,并用硫酸盐对其改性,极大地提高了噻吩类硫化物的脱除率。J.González等[12]以乙腈为萃取剂,双氧水为氧化剂,25%⁃V2O5/Ti⁃MCM⁃41为催化剂构建氧化脱硫体系,该体系对模拟油中的二苯并噻吩具有较好的脱除效果。C.Wang等[13]以SBA⁃15为载体,通过浸渍法合成了V2O5/SBA⁃15催化剂,以氧气为氧化剂,并以离子液体作为萃取剂应用于油品的氧化脱硫,取得了良好的深度脱硫效果。石墨相氮化碳(g⁃C3N4)作为一种半导体材料,因其具有成本低、化学稳定性高、催化活性高的特点,成为一种优良的载体[14]。g⁃C3N4与V2O5复合后的V2O5/g⁃C3N4催化剂在光催化领域有着重要的应用。Q.Liu等[15]采用一步法制备了V2O5/g⁃C3N4复合材料,在可见光下,以罗丹明B为降解物,发现其具有很高的光催化活性。T.Jayaraman等[16]以V2O5和g⁃C3N4为原料,采用湿浸渍法制备了V2O5/g⁃C3N4纳米复合材料,并对其光降解能力进行研究。Y.Hong等[17]通过原位生长的方式制备了Z型V2O5/g⁃C3N4异质结光催化剂,在可见光下对罗丹明B和四环素进行降解研究。这一系列研究表明,V2O5/g⁃C3N4催化剂在光催化领域备受关注,但是其在氧化脱硫方面的研究却很少见。
本文以偏钒酸铵、三聚氰胺、硼酸为前驱体,通过高温煅烧法制备V2O5/g⁃C3N4催化剂,方法简单,活性组分V2O5在氮化碳上原位合成,分散均匀。以V2O5/g⁃C3N4为脱硫催化剂,乙腈为萃取剂,双氧水为氧化剂构成脱硫体系。实验考察了反应温度、氧硫物质的量比、催化剂质量、萃取剂体积以及不同硫化物等因素的影响,并对其脱硫机理进行探讨。
1 实验部分
1.1 试剂
无水甲醇,分析纯,天津市瑞金特化学品有限公司;偏钒酸铵,分析纯,天津光复精细化工研究所;硼酸,分析纯,天津市大帽化学试剂厂;三聚氰胺(分析纯)、正辛烷(纯度98%)、H2O2(质量分数30%),国药集团试剂有限公司;二苯并噻吩(DBT,纯度98%)、4,6二苯并噻吩(4,6⁃DMDBT,纯度97%)、苯并噻吩(BT,纯度99%),阿拉丁试剂有限公司。
1.2 仪器
WK⁃2D微库仑综合分析仪,江苏江分电分析仪器有限公司;NEXUS 870傅里叶变换红外光谱仪,美国尼高力仪器公司;D8 Advance Bruker型X射线衍射仪,德国布鲁克光谱仪器公司;JSM⁃6610LV扫描电子显微镜,日本电子株式会社;ASAP2010N2吸附⁃脱附等温仪,美国麦克仪器公司。
1.3 V2O5/g⁃C3N4催化剂的制备
称取0.60 g偏钒酸铵,置于马弗炉中,在氮气保护下530℃煅烧3 h,得到样品V2O5。称取4.00 g三聚氰胺和1.47 g硼酸溶解于30 mL无水甲醇中,并在磁力搅拌器上搅拌30 min;置于加热套中蒸出甲醇,得到白色固体;然后将固体烘干,研磨后煅烧3 h,得到样品g⁃C3N4。称取4.00 g三聚氰胺、0.60 g偏钒酸铵和1.47 g硼酸溶解于30 mL无水甲醇中,搅拌一段时间后蒸出甲醇,然后经烘干、煅烧后得到样品V2O5/g⁃C3N4。
1.4 氧化脱硫实验
将0.718 g DBT超声溶解于500 mL正辛烷中,得到质量浓度为250 mg/L的DBT模拟油。在带有冷凝装置的三角瓶中加入5.0 mL模拟油和一定量的乙腈、H2O2、催化剂,然后置于恒温水浴锅中搅拌并反应60 min,每隔一段时间取少量上层油相。用WK⁃2D微库仑综合分析仪测定硫的质量浓度,并计算脱硫率,其公式为:
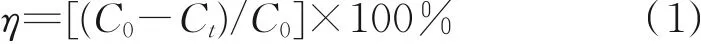
式中,η为反应的脱硫率,%;C0和Ct分别为初始和t时刻模拟油硫质量浓度,mg/L。
2 结果与讨论
2.1 XRD表征
以偏钒酸铵和三聚氰胺为V2O5和g⁃C3N4的前驱体,通过高温煅烧制备了纳米复合材料V2O5/g⁃C3N4。由于过高的温度和过长的反应时间可能会使g⁃C3N4分解,所以利用X⁃射线粉末衍射技术考察催化剂的制备是否成功。图1为不同样品的XRD谱图。从图1可以看出,g⁃C3N4的谱图中27.3°处有很强的特征衍射峰对应(002)晶面,12.9°的特征衍射峰为(100)晶面,说明g⁃C3N4与石墨具有类似的层状结构,与文献[18−19]报道一致。V2O5的特征衍射峰为15.4°、20.3°、21.6°、26.1°、31.0°,分别对应V2O5的(200)、(010)、(110)、(101)、(310)晶 面[20],与V2O5标准谱图(JCPDS#45⁃10741)的特征衍射峰相吻合,属于正交晶型。合成样品的主要峰强且尖锐,说明样品的结晶度较好。V2O5/g⁃C3N4样品中V2O5特征衍射峰与纯V2O5特征衍射峰相比变弱,g⁃C3N4的特征衍射峰向右偏移,表明部分V2O5粒子可能进入到g⁃C3N4层状结构内部[21]。上述结果表明,通过煅烧偏钒酸铵和三聚氰氨等前驱物可以合成V2O5/g⁃C3N4。
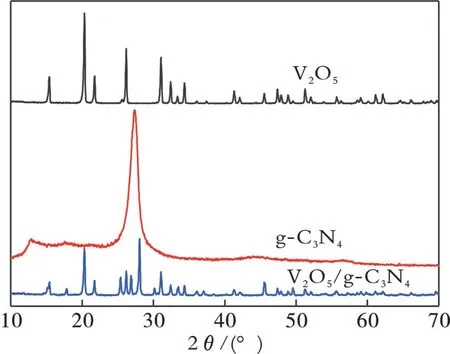
图1 不同样品的XRD谱图Fig.1 XRD spectrum of different samples
2.2 FT⁃IR表征
为了进一步验证催化剂的合成,采用红外光谱对催化剂的结构进行分析。图2为不同样品的红外光谱。从图2可以看出,3 406、3 193 cm−1处对应的宽峰为N−H伸缩振动峰;在g⁃C3N4的红外谱图中,1 246~1 635 cm−1处对应的吸收峰为C=N和芳香C−N杂环分子伸缩振动[22],802 cm−1处对应的特征吸收峰是由三嗪环结构的面外振动引起的[16];在V2O5光谱图中,1 024 cm−1处吸收峰为V=O伸缩振动,810 cm−1处吸收峰为V−O−V伸缩振动。与文献[15]相比,V2O5的红外谱图在560 cm−1处出现新的吸收峰,这可能源于V2O5中的晶格氧[23]。据报道,钒氧化合物中的晶格氧可参与噻吩类的脱硫反应,使催化剂具有更好的催化氧化性能[13]。复合后,在V2O5/g⁃C3N4的红外谱图中均能看到V2O5与g⁃C3N4的吸收峰,未检测出其他杂质吸收峰,这表明钒的氧化物已经成功负载到g⁃C3N4上,该结论与XRD的表征一致。
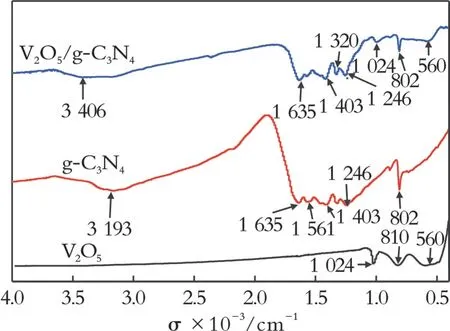
图2 不同样品的红外光谱Fig.2 FT⁃IR spectra of different samples
2.3 XPS表征
为了进一步确认催化剂元素组成和活性组分的价态形式,对V2O5/g⁃C3N4进行XPS分析,结果见图3。从图3可以看出,复合样品中检测到C、N、O和V等元素;在g⁃C3N4的C1s谱图中存在284.6、288.0、286.2 eV三个特征峰[17],其中特征峰为284.6 eV归属于C−C,288.0 eV归属于N−C=N,而286.2 eV处的特征峰可能是表面吸收氧而形成的C−O[24]。在g⁃C3N4的N1s谱图中,398.3 eV处有一个特征峰,为N原子与两个C原子的sp2(C=N−C)杂化[25],而复合后C、N的结合能变为398.7 eV,这表明g⁃C3N4与V2O5发生了相互作用;在g⁃C3N4的V2p谱图中,517.2 eV与524.8 eV附近观察到V2p3/2和V2p1/2对应的V2O5峰[13,17],且未发现V4+存在,因此可以判断V元素在g⁃C3N4上分布是以V2O5形式存在;原V2O5的O1s谱图中在529.8 eV处特征峰,复合后出现在532.4 eV处,这可能与正交V2O5中的O2−有关[26]。

图3 V2O5/g⁃C3N4的XPS分析Fig.3 XPS spectrum of V2O5/g⁃C3N4
2.4 SEM表征
图4 为不同样品的SEM图。从图4(a)可以看出,g⁃C3N4为带有大量空隙的片状结构聚集体,样品中空隙是煅烧过程中裂解气体造成的,使样品更加多孔,比表面积有所增大。从图4(b)可以看出,V2O5为块状结构,尺寸较大。从图4(c)可以看出,已形成V2O5与g⁃C3N4的混合体,这表明通过直接煅烧法可以合成V2O5/g⁃C3N4。

图4 不同样品的SEM图Fig.4 SEM images of different samples
2.5 BET表征
对于负载型催化剂,较大的比表面积和孔径有利于分散活性组分和减小扩散阻力,进而提高催化剂的活性。因此,考察催化剂的比表面积与孔隙结构参数,结果见表1。
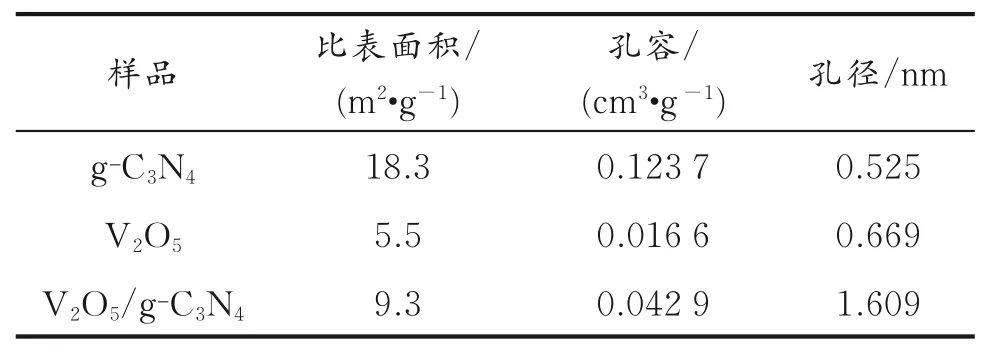
表1 不同的样品的比表面积和孔隙结构参数Table 1 Specific surface area and pore structure parameters of different samples
由表1可 知,g⁃C3N4、V2O5的比表面 积 分别为18.3、5.5 m2/g,负 载 后 的 比 表 面 积 为9.3 m2/g。V2O5/g⁃C3N4与g⁃C3N4相比,比表面积与孔容下降,这是因为部分V2O5颗粒填充g⁃C3N4孔道,孔径增加则是因为V2O5颗粒在g⁃C3N4表面聚集堆积的结果[28]。
图5 为不同样品的N2吸附⁃脱附等温线。
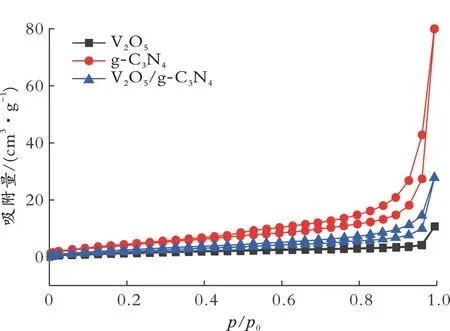
图5 不同样品的N2吸附⁃脱附等温线Fig.5 N2 adsorption⁃desorption isotherms of different samples
从图5可以看出,V2O5/g⁃C3N4和g⁃C3N4的吸附⁃脱附曲线中均出现回滞环,属于第Ⅳ类吸附曲线,这说明负载后的V2O5/g⁃C3N4的催化剂是介孔材料[27]。
2.6 氧化脱硫反应条件的优化
2.6.1 不同脱硫体系对脱硫率的影响 不同脱硫体系对DBT的脱除率具有不同影响,分别考察了V2O5、g⁃C3N4和V2O5/g⁃C3N4等对模拟油中DBT的脱除效果的影响,结果见表2。由表2可知,g⁃C3N4的吸附脱硫率仅为2.7%。以V2O5为催化剂,乙腈为萃取剂,H2O2为氧化剂,其脱硫率为76.8%;而以V2O5/g⁃C3N4为催化剂,在相同条件下脱硫率达到91.9%,这表明复合后的V2O5/g⁃C3N4催化剂对DBT的脱除效率明显提高。
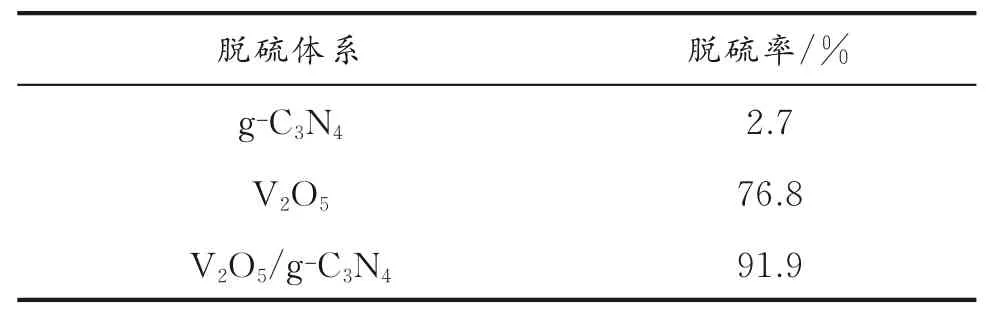
表2 不同脱硫体系对脱硫率的影响Table2 Influence of different desulfurization systems on removal of sulfur compounds
2.6.2 温度对脱硫率的影响 在模拟油体积为5 mL、乙腈体积为1.5 mL、催化剂质量为0.020 g、n(H2O2)/n(S)为8的条件下,考察温度对脱硫率的影响,结果见图6。从图6可以看出,当反应时间为60 min时,温度从20℃增加至30℃,脱硫率从72.9%增加至77.3%,继续升高温度,脱硫率反而下降。这是因为反应温度升高,双氧水的分解加剧[29],同时金属离子促进双氧水分解[30]。综合脱硫效果和节能方面的考虑,最佳温度为30℃。
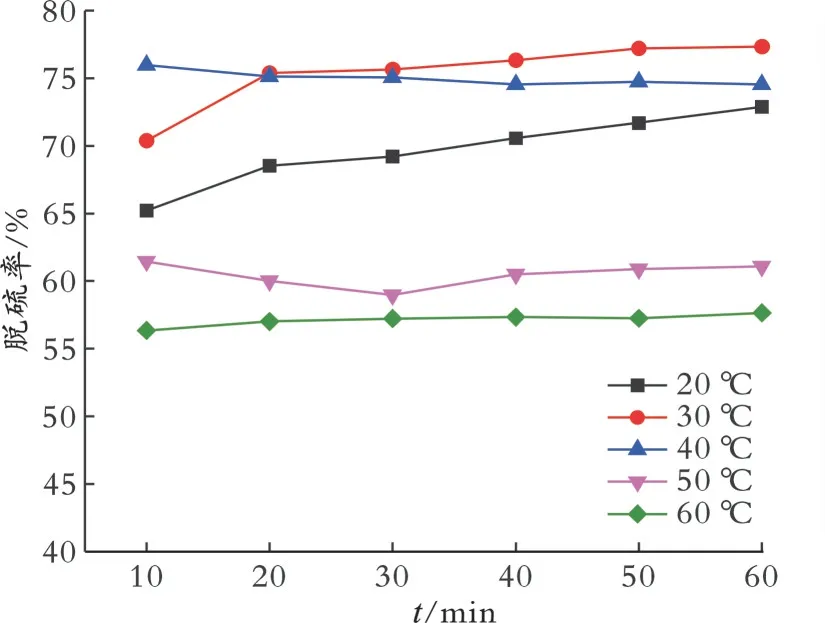
图6 温度对脱硫率的影响Fig.6 Influence of the reaction temperature on desulfurization rate
2.6.3 催化剂质量对脱硫率的影响 在模拟油体积为5.0 mL、乙腈体积为1.5 mL、n(H2O2)/n(S)为8、温度为30℃的条件下,考察催化剂质量对脱硫率的影响,结果见图7。从图7可以看出,当反应时间为60 min时,催化剂质量从0.010 g增加至0.020 g,脱硫率从40.2%增加至77.3%,继续增加催化剂质量,脱硫率反而下降。这是因为催化剂质量增加会增加活性位的数量,但是催化剂质量继续增加时,双氧水与DBT的竞争吸附导致脱硫率下降[31]。因此,最佳催化剂质量为0.020 g。

图7 催化剂质量对脱硫率的影响Fig.7 Influence of the catalyst quality on desulfurization rate
2.6.4n(H2O2)/n(S)对脱硫率的影响 根据化学反 应计量 比,2 mol的H2O2可 以将1 mol DBT氧化为DBTO2。但是,由于存在双氧水的分解反应,通常双氧水的加入量会大大增加[32]。在模拟油体积为5.0 mL、乙腈体积为1.5 mL、催化剂的质量为0.020 g、温度为30℃的条件下,考察n(H2O2)/n(S)对脱硫率的影响,结果见图8。从图8可以看出,当反应时间为60 min、n(H2O2)/n(S)=2时,脱硫率为53.4%;相同的反应时间下,n(H2O2)/n(S)继续增加至4、6、8时,脱硫率增加,分别为59.2%、72.8%、73.4%;n(H2O2)/n(S)继续增加至10时,脱硫率无明显变化。因此,最佳n(H2O2)/n(S)=8。
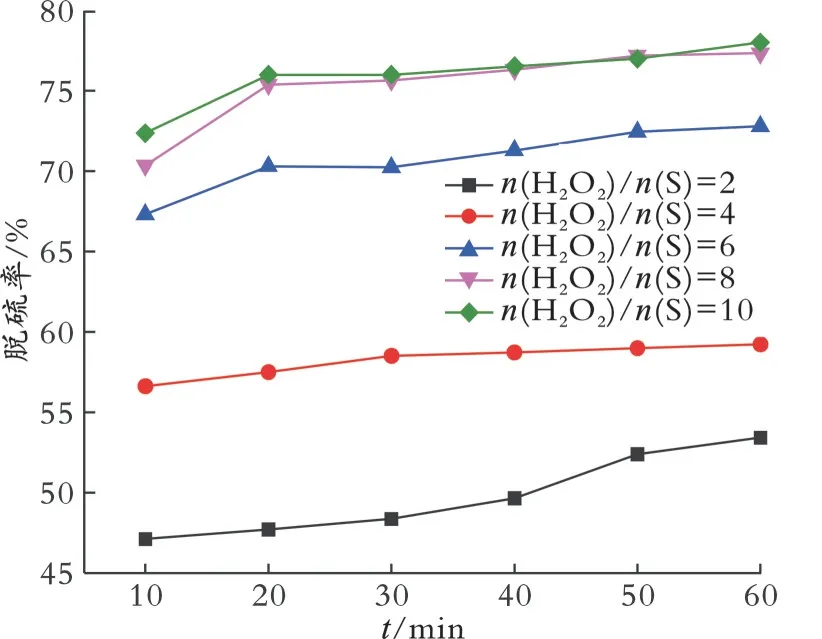
图8 n(H2O2)/n(S)对脱硫率的影响Fig.8 Influence of n(H2O2)/n(S)on desulfurization rate
2.6.5 萃取剂体积对脱硫率的影响 在模拟油体积为5.0 mL、催化剂质量为0.020 g、温度为30℃、n(H2O2)/n(S)为8的条件下,考察萃取剂乙腈体积对脱硫率的影响,结果见图9。从图9可以看出,当反应时间为60 min时,萃取剂体积从1.5 mL增加至3.0 mL时,脱硫率从77.3%增加至91.9%,继续增加萃取剂体积,脱硫率略有降低。这是因为增加萃取剂体积使更多的DBT进入萃取相中被氧化成相应的砜类物质,从而提高脱硫率[33]。但是,由于双氧水的加入量一定,当较多的硫化物被萃取到乙腈相中后,脱硫体系的氧硫物质的量比也随之降低,脱硫效果会变差[34]。因此,最佳萃取剂体积为3.0 mL。
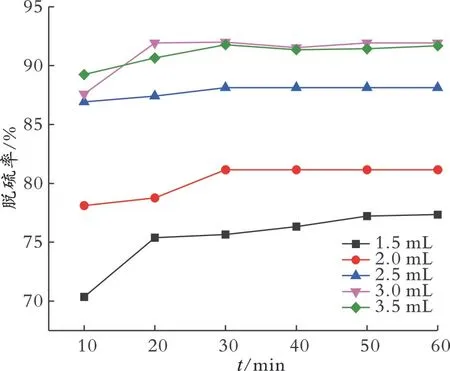
图9 萃取剂体积对脱硫率的影响Fig.9 Influence of the extractant volume on desulfurization rate
2.6.6 不同硫化物对脱硫率的影响 催化剂对不同硫化物具有不同的选择性,在模拟油体积为5.0 mL、乙腈体积为3.0 mL、催化剂质量为0.020 g、温度为30℃、n(H2O2)/n(S)为8的条件下,考察了催化剂对不同硫化物的脱除情况,结果见图10。从图10可以看出,脱硫顺序为DBT(91.9%)>BT(68.0%)>4,6⁃DMDBT(47.9%),其差异可能与它们的电子云密度和硫原子周围空间位阻有关。DBT、4,6⁃DMDBT和BT硫原子上的电子密度分别为5.758、5.760和5.739。随着电子云密度的增加,脱硫效果逐渐增加[35]。但是,4,6⁃DMDBT因其结构上存在两个甲基,空间位阻大,阻碍了硫原子与V2O5/g⁃C3N4催化剂活性位的接触,导致脱硫率最低。可知,催化剂经过5次循环,脱硫率仍然可以达到85.7%,说明催化剂具有良好的稳定性。随着回收次数的增多,催化剂脱硫率降低,其原因可能是催化剂活性组分的流失。
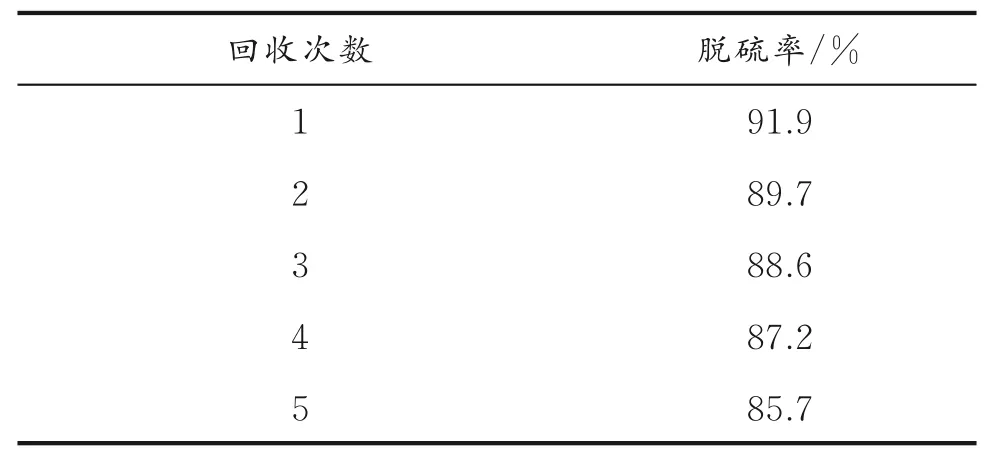
表3 催化剂的回收次数对脱硫率的影响Table 3 Effect of catalyst recovery on desulfurization rate

图10 催化剂对不同硫化物的脱除情况Fig.10 Removal of different sulfides by catalyst
2.6.7 催化剂的回收次数对脱硫率的影响 催化剂经催化氧化脱硫后,用分液漏斗移去上层油相,采用离心法分离出固相的催化剂,该催化剂用去离子水和无水乙醇洗涤、干燥。在模拟油体积为5.0 mL、乙腈体积为3.0 mL、催化剂质量为0.020 g、温度为30℃、n(H2O2)/n(S)为8的条件下,考察催化剂的回收次数对脱硫率的影响,结果见表3。由表3
2.7 催化剂脱硫反应机理
V2O5/g⁃C3N4催化氧化脱硫机理如图11所示。在该脱硫体系中起主要作用的是催化中心V2O5,g⁃C3N4则为活性组分提供更大的比表面积,使活性组分分散均匀而形成更多活性位点。
在一定量模拟油中加入乙腈与V2O5/g⁃C3N4催化剂后,该氧化脱硫体系分为上下两相,即上层的模拟油相和下层包含催化剂的乙腈相[35⁃36]。研究表明,V2O5在双氧水的作用下会形成过氧化物具体过程如下:首先,油相中的DBT被萃取到乙腈相中,与发生亲核取代中的活性氧转移到S原子上,DBT被氧化为亚砜,并进一步生成砜。H2O2不断地将转化为,从而形成催化循环,以保证脱硫反应正常进行,直至双氧水耗尽[39]。

图11 V2O5/g⁃C3N4催化氧化脱硫机理Fig.11 Proposed mechanism for V2O5⁃catalyzed oxidation of DBT
3 结论
(1)采用煅烧法制备了V2O5/g⁃C3N4,通过XRD证明部分V2O5进入到g⁃C3N4的片状结构中。XPS谱图中观测到四种元素,且在FT⁃IR谱图中未观测到其余杂质峰,这表明成功制备了V2O5/g⁃C3N4催化剂。
(2)以V2O5/g⁃C3N4为催化剂,乙腈为萃取剂,双氧水为氧化剂,考察了反应条件对模拟油中硫化物脱除率的影响,在最优脱硫条件下对DBT的脱除率可达91.9%。
(3)对V2O5/g⁃C3N4重复利用实验表明,V2O5/g⁃C3N4在经5次再生后对DBT的脱除率可以达到85.7%,仍具有较高的催化活性。
