制药用压缩空气制备系统及质量标准分析
2021-04-02赵树勇

摘要:制药生产技术日新月异,洁净压缩空气的应用范围逐步拓宽,成为药品制造领域的关键工艺源气,由于其直接与原辅料及内包装接触,因此将显著影响药品的品质。现对制药用压缩空气制备系统及质量标准展开分析,涉及悬浮微粒、微生物限度等各类用于评价空气洁净度的关键指标,并提出现阶段业内主流的制备技术,可供同仁参考。
关键词:洁净压缩空气;空气制备;悬浮微粒
1 药用洁净压缩空气质量指标
洁净度是药品生产中的关键控制对象,也是评价药品质量的重要指标,原料药精制、烘干等一系列生产活动均需在洁净环境中完成。其中,空调净化系统能给药品质量的控制提供帮助,用于调控药品生产环境,保证空气温湿度、悬浮粒子等指标均稳定在许可范围内,由此规避空气污染和交叉污染,给药品的品质保驾护航[1-2]。对于以注射剂、眼用制剂为代表的各类制剂,其临床应用途径普遍特殊,需要直接注入特定的器官和血液系统或用于外伤患者的创口处,为最大限度减小对患者的不良影响,必须保证制剂的无菌性。此外,从输液制剂的角度来看,其安全性风险的决定性因素较多,除原材料的品质外,还与微生物、细菌内毒素等物质的含量存在密不可分的关系。对于前述所提的各类无菌制剂的生产,均发生在洁净度级别在C级以上的环境中,部分核心环节需提高至A级标准,所用的洁净压缩空气需得到深度的净化和无菌处理,而洁净压缩空气系统则是实现此类目标的关键“参与者”。
2 洁净压缩空气系统设计及技术控制
2.1 洁净压缩空气制备系统的运行工艺流程
以空气压缩机、干燥及过滤净化装置为核心,结合各类辅助设备共同组成洁净压缩空气制备系统。实际运行中以药品剂型对压缩空气的要求为导向,采取合适的工艺流程,主体内容有:吸气过滤器→空气压缩机→冷却器→储气罐→前置过滤器→精密过滤器→干燥装置→后置过滤器→除味过滤器→除菌过滤器→输送管网。
所提流程并非一成不变,而需根据实际情况灵活配置,重点考虑生产规模、药品剂型对压缩空气的要求(用量、洁净度等方面),由此提高压缩空气净化工艺流程的可行性。
以GMP要求及现行的药品剂型质量要求为基准,结合历史经验,推荐采用图1所示的洁净压缩空气制备系统。
2.2 空气压缩机、干燥设施及过滤器的配置
(1)空气压缩机。制药领域的空气压缩机主要有三大类:一是离心式,单台设备容量较大,在小气量运行过程中容易出现喘振现象,易影响制药的连续性和药品的品质,因此不推荐;二是活塞式,设备规格较大,运行期间伴有明显的噪声污染和振动问题,因此也不推荐;三是螺杆式,此类空气压缩机结构紧凑,零部件配合效果较佳,自动化水平较高,可作为空气压缩机的首选类型。
(2)干燥设施。冷冻式和吸附式是两种较为典型的压缩空气干燥方式,选择时主要以压缩空气的压力露点为判断依据。压力露点超过3 ℃时,推荐采用冷冻式干燥机,其运行稳定、流量范围较大,且全程几乎不存在压缩空气被无故消耗的情况;若压力露点在3 ℃以内,则推荐采用吸附式干燥机,或是在此基础上增设冷冻式干燥机,形成联合作业的模式。
(3)过滤器。过滤器的主要作用在于滤除空气中的无用物质,例如微生物、尘埃粒子颗粒物。现阶段,精密过滤器、活性炭过滤器等均是较为典型的产品,各自的应用特点存在差异,需根据实际需求合理选择。
2.3 洁净压缩空气制备系统设计的技术要点
(1)缓冲罐的配置。在制药规模逐步扩大的背景下,制药领域普遍使用大中型压缩空气系统,其存在较丰富的用气点,各自的用气量不尽相同,在用气量高峰期易由于需求的增加而出现系统压力大幅波动的情况。若压力偏低,受压力波动的影响过滤器易受损,难以保证压缩空气的洁净度。为解决此问题,需在空气压缩机后方的工艺链上设置储气罐。而对于压力波动小、洁净度要求高的情况,有必要根据洁净区的实际特点适配不锈钢平衡罐,且需要根据该装置的特点合理配置呼吸器。
(2)工藝源气和仪表用气的设置常采取的是合建1个空气站的方式,以最高用气压力为准合理调控系统的供气压力,在终端过滤器前增设减压阀,利用该装置满足较低用气点的压力需求。
(3)输送管网配套工作中,阀门和管路两部分均由不锈钢材料制得,其中以316L型不锈钢最为合适,焊接推荐采用氩弧焊的方法。
(4)若配套的是吸附式干燥机,则需在上游端增设除油过滤器,利用该装置滤除空气的油污,以免干燥剂的品质受到影响。此外,在下游端配套后置过滤器,以达到消除干燥剂粉尘的效果。应用冷冻式干燥机时,必须在工艺链的上游配套前置过滤器。
3 实例检测
3.1 医药级洁净压缩空气制备装置的配套方案
实例1:根据图1(a)(灭菌及无菌制剂用)所示方式,按顺序依次配置设备,其中前置过滤器的过滤孔径为5 μm,第一、第二精密过滤器的过滤孔径为1 μm、0.1 μm,活性炭过滤器的过滤孔径为0.01 μm。在将空气引入制备系统后,调控空气压缩机输出的压缩空气的压力,使其稳定在0.78~0.96 MPa,再由冷冻式干燥机输出温度为30 ℃的压缩空气,在此期间吸附式干燥器运行,输出压力露点为-42 ℃的压缩空气,经各类设备的一系列处理后由终端过滤器输出气体,以便给制药阶段提供洁净压缩空气。
实例2:按实例1的方式配备设备,但前置过滤器、第一精密过滤器的过滤孔径分别调整为4 μm、0.5 μm。在将空气引入制备系统后,调控空气压缩机输出的压缩空气的压力,使其稳定在0.78~0.96 MPa,再由冷冻式干燥机输出温度为28 ℃的压缩空气,在此期间吸附式干燥器运行,输出压力露点为-42 ℃的压缩空气,经各类设备的一系列处理后由终端过滤器输出气体,依然可提供洁净压缩空气。
实例3:按实例2的方式配置设备,在将空气引入制备系统后,调控空气压缩机输出的压缩空气的压力,使其稳定在0.80~0.95 MPa,再由冷冻式干燥机输出温度为26 ℃的压缩空气,在此期间吸附式干燥器运行,输出压力露点为-45 ℃的压缩空气,经各类设备的一系列处理后由终端过滤器输出气体,依然可提供洁净压缩空气。
实例4:按实例1的方式配备设备,但前置过滤器、第一精密过滤器、终端过滤器的过滤孔径分别调整为3 μm、0.22 μm、0.1 μm。在将空气引入制备系统后,调控空气压缩机输出的压缩空气的压力,使其稳定在0.80~0.90 MPa,再由冷冻式干燥机输出温度为26 ℃的压缩空气,在此期间吸附式干燥器运行,输出压力露点为-45 ℃的压缩空气,经各类设备的一系列处理后由终端过滤器输出气体,依然可提供洁净压缩空气。
在落实前述4种方案后,分别对制备的医药级洁净压缩空气进行取样,并展开测定与分析工作。
3.2 檢测项目、方法及结果
(1)组织含油量测试,在压缩空气终端用气点取样,所用仪器为专用采样管,按规范测定。
(2)组织可见异物和细菌内毒素测试,在压缩空气终端用气点取样,向密封瓶内注入压缩空气,在此期间调控压力并使其维持稳定,使气体从瓶底逐步通过灭菌注射用水。测试人员用专用采样器测定流量数,在此期间密切观察通气量,待其增加至设定值后即可取出采样器,对采样瓶采取密封措施,用于检验(采用细菌内毒素检查法)。
(3)组织尘埃粒子测试,在压缩空气终端用气点取样,将5 L锥形瓶直立,调节压缩空气的压力使其可通入瓶中,待瓶内空气被完全排出后利用尘埃粒计数器测定。
(4)组织微生物数测试,在压缩空气终端用气点取样,用减压阀与压缩空气取样装置连接,并连接采样装置和浮游菌采样仪,由此展开检测(遵循浮游菌检测规程)。
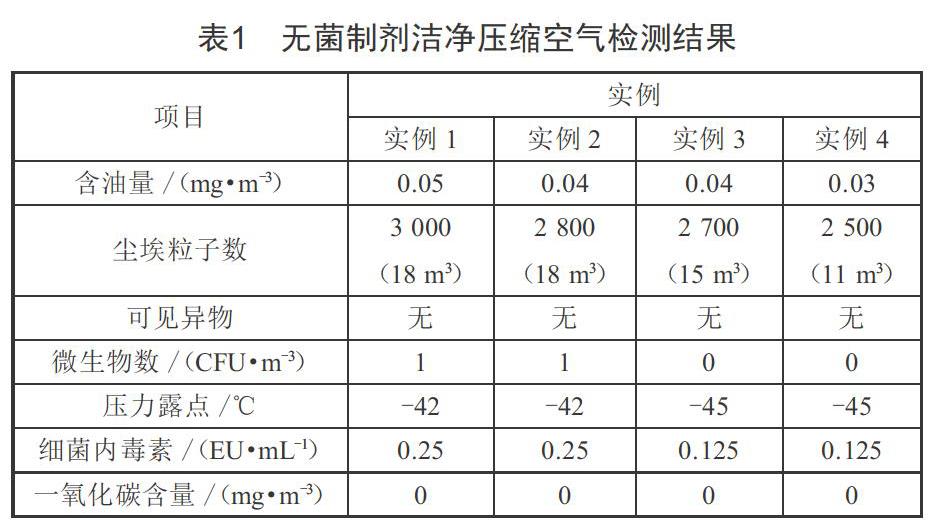
(5)组织压力露点测试,在压缩空气终端用气点取样,所用仪器为压缩空气露点仪。汇总所得结果,如表1所示。
由表1可知,在采取前述所提4种配置方案后,所产生的无菌制剂洁净压缩空气各项指标均可满足要求,可直接接触药品,能够保证药品生产品质。
4 结语
为满足药品生产品质要求,制药阶段需配套压缩空气系统,为充分发挥出该系统的应用优势,在实际工作中,工作人员需根据药品制剂对压缩空气的要求合理配套设备,切实提高质量标准,保证压缩空气系统可正常运行,给药品生产提供支持。
[参考文献]
[1] 任红兵.药用纯化水制备技术及装备发展研究[J].机电信息,2015(26):1-12.
[2] 朱莲华.制药生产所用压缩空气的认识性探讨[J].机电信息,2011(8):41-46.
收稿日期:2021-01-12
作者简介:赵树勇(1976—),男,广东潮州人,工程师,从事生物制药动力系统、冷库、暖通等方面的工作。