Mechanism and selectivity of copper-catalyzed borocyanation of 1-aryl-1,3-butadienes: A computational study
2021-04-02XinzhiLiXiangweiRenHongliWuWentaoZhaoXiangyangTangGenpingHuang
Xinzhi Li,Xiangwei Ren,Hongli Wu,Wentao Zhao,Xiangyang Tang*,Genping Huang*
Department of Chem istry, School of Science and Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Tianjin University, Tianjin 300072, China
ABSTRACT Density functional theory calculations have been performed to investigate the copper-catalyzed borocyanation of 1-aryl-1,3-butadienes.The computations show that the regio-and enantioselectivity is determined by the borocupration step.The π-electron withdrawing aryl group at the C1 atom makes the C4 atom more electrophilic than the other carbon atoms,which together with the steric repulsion around the forming C--B bond,results in the experimentally observed exclusive 4,3-regioselectivity.The origins of the enantioselectivity were attributed to the steric effect and π-π stacking interaction between the butadiene moiety and the ligand.
Keywords:Copper catalyst Borocyanation Mechanism Selectivity DFT calculations
The boron-containing compounds are widely observed in medicinal chemistry and material science[1],and are key building blocks in a range of organic transformations[2].The development of the efficient strategies for the synthesis of these compounds is thus of vital importance in modern organic synthesis[3,4].In this regard,the copper-catalyzed borylative functionalizations of C--C multiple bonds, which represents one of the most efficient and straightforward protocols, have attracted extensive research interest over the past two decades [4,5].From a mechanistic point of view, the key step of these reactions is the initial borocupration of the C--C multiple bonds to afford the organocopper species, which then can react with the electrophilic coupling partners to produce the final boron-containing compounds [6].
Despite the significant progress has been made in this field,the development of the regio-and enantio-selective reactions remains a key challenge.In this context, Procter and co-workers recently reported an elegant example of the Cu-catalyzed borocyanation of 1-aryl-1,3-butadienes under mild reaction conditions (Scheme 1)[7].It was found that with the in situ generated LCu-Bpin complex,the reactions of the 1-aryl-1,3-butadienes 1 with the N-cyano sulfonamide 2 can give the borocyanation products(S)-3 with the complete 4,3-regioselectivity and good enantioselectivity (up to 93%ee).The reaction thus represents a rare case of the regio-and enantio-selective borylative functionalization of unsymmetrical 1,3-dienes.
Considering the origins of the selectivity remain unclear and following our continuous research interest in this field [8], we therefore decided to investigate the title reaction by means of density functional theory (DFT) calculations (see Supporting information for the computational details).The results show that the selectivity of the overall reaction is determined by the borocupration step,and the origins of the experimentally observed regio-and enantio-selectivities are attributed to the combination of the electronic and steric effects.The present results, together with our previous work[8a],may provide important implications for a better understanding of related Cu-catalyzed borylative functionalization reactions.
The 1-phenyl-1,3-butadiene 1a was selected as the model substrate in the current calculations.The calculated energy profile of the 4,3-borocyanation of 1a is given in Fig.1.The computations show that the reaction is initiated by the coordination of the C3-C4double bond of 1a to the Cu center of active catalyst A, leading to the formation of intermediates (S)-INT1 and (R)-INT1, which was calculated to be exergonic by 2.2 and 1.2 kcal/mol, respectively.Then, the ensuing 4,3-borocupration was found to take place via transition states(S)-TS1 and(R)-TS1,with the energy barriers of 7.2 and 9.5 kcal/mol relative to (S)-INT1.The borocupration was calculated to be highly exergonic,as evidenced by the resulted allyl copper intermediates (S)-INT2 and (R)-INT2 being much more stable than (S)-INT1 by 22.0 and 24.3 kcal/mol, respectively.
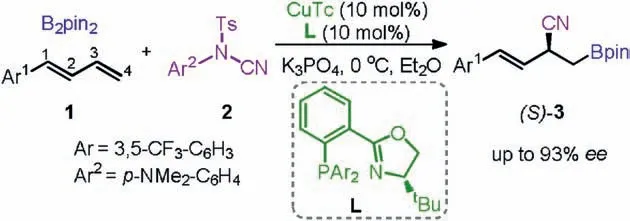
Scheme 1.Copper-catalyzed borocyanation of 1-aryl-1,3-butadienes 1.
From intermediates (S)-INT2 and (R)-INT2, the electrophilic cyanation can occur via the migratory insertion of the C≡N triple bond of N-cyano sulfonamide 2 into the Cu-C3bond.However,this possible pathway was calculated to be kinetically infeasible,requiring energy barriers of higher than 30 kcal/mol (see Supporting information for details).Instead, we found that (S)-INT2 and (R)-INT2 can first undergo an η1-allyl isomerization via transition states(S)-TS2 and(R)-TS2 to give intermediates(S)-INT3 and (R)-INT3 [9].Subsequently, the electrophilic cyanation at C3position takes place via six-membered transition states(S)-TS3 and(R)-TS3, giving the intermediates (S)-INT4 and (R)-INT4, respectively.The computations show that the electrophilic cyanation via the six-membered transition states is much more favorable than that via the four-membered migratory insertion transition states(see Supporting information for other possibilities), being in accordance with the previous computational studies [6c].
Instead of the η1-allyl isomerization,the electrophilic cyanation at C1position can occur directly from (S)-INT2 and (R)-INT2 via transition states (S)-TS3′and (R)-TS3′(Scheme 2).However, this possibility can be ruled due to the high energy barriers required compared with those of(R)-TS3 and(R)-TS3,being in line with our previous study [8a].The results are thus in accordance with the experimental results that no cyanation at C1position was observed[7].The origins are probably due the steric repulsion around the forming C--C bond in (S)-TS3′and (R)-TS3′is larger than that in(S)-TS3 and (R)-TS3.
The next step of the reaction is the 1,2-elimiantion of the copper tosylamide INT5, which was calculated to proceed via transition states (S)-TS3 and (R)-TS3, leading to the 4,3-borocyanation products (S)-3a and (R)-3a, respectively.
The catalytic cycle is finally completed by the regeneration of the active catalyst A with the assistance of K3PO4.The computations show that the ligand exchange between INT5 and K3PO4to give intermediates INT6 and INT7 is exergonic by 7.9 kcal/mol.Then,the incoming B2pin2binds to intermediate INT7 to generate intermediate INT8, from which the transmentallation via transition state TS5 is quite facile, with an energy barrier of only 2.8 kcal/mol relative to INT8.The transmentallation process was calculated to be highly exergonic, and the resulting intermediate INT9 is more stable than INT8 by as much as 19.1 kcal/mol.The active catalyst A can be regenerated via the release of K2PO4-Bpin from INT9.
The results show that the borocupration constitutes the rate-and selectivity-determining step of the overall reaction.The overall energy barrier of the reaction is 21.7 kcal/mol,which was calculated by the energy difference between INT9 and A (16.7 kcal/mol) and the energy difference between A and (S)-TS1 (5.0 kcal/mol).The calculated energy difference of 2.3 kcal/mol(5.0 kcal/mol of(S)-TS1 versus 7.3 kcal/mol of (R)-TS1) corresponds to a calculated enantioselectivity of around 97%ee at 0°C,which is in agreement with the experimentally observed 91%ee[7].
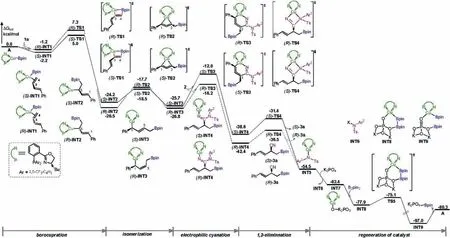
Fig.1.Calculated energy profile of the Cu-catalyzed 4,3-borocyanation of 1a.
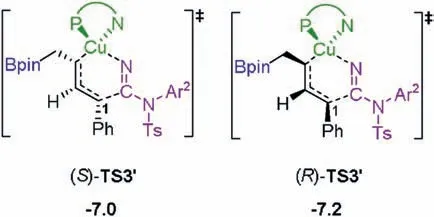
Scheme 2.Electrophilic cyanation at C1 position.
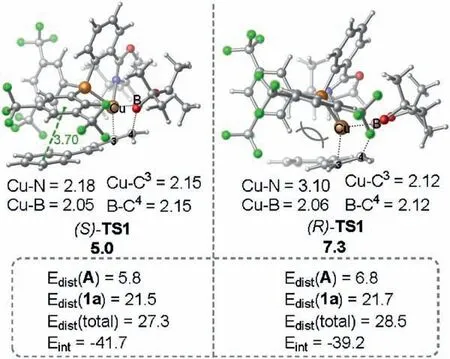
Fig.2.Optimized geometric structures of (S)-TS1 and (R)-TS1.The distortion/interaction energies were calculated at the level of ωB97XD-SMD(Et2O)/6-311+G( d,p)-SDD.The energies and selected bond distances are given in kcal/mol and Å,respectively.
The origins of the enantioselectivity are attributed to both electronic and steric effects.Fig.2 shows the optimized geometries of the enantioselectivity-determining transition structures(S)-TS1 and (R)-TS1.It was found that in transition state (R)-TS1, the butadiene moiety is oriented toward to the bulky tBu group of the oxazoline ring.As such, the steric repulsion between the butadiene moiety and the oxazoline ring can be expected,whereas no such steric repulsion exists in (S)-TS1.To minimize the steric repulsion,the oxazoline ring in (R)-TS1 was found to be distorted(Cu-N=3.10 Å in (R)-TS1 versus Cu-N=2.18 Å in (S)-TS1).Furthermore, since that the butadiene moiety is oriented toward to the phosphorus moiety of the ligand in (S)-TS1, the π-π stacking interaction between the phenyl group of 1a and the aryl ring on the phosphorus atom was observed (3.70 Å).Therefore, the combination of the steric and electronic effects makes transition state(S)-TS1 being more favorable than transition state(R)-TS1,enabling the experimentally observed enantioselectivity.The distortion/interaction analysis[10]of(S)-TS1 and(R)-TS1 also shows that the distortion energy of active catalyst A (Edist(A)) and interaction energy (Eint) in (S)-TS1 are both more favorable than those in(R)-TS1, providing thus further support for our argument.
Finally, apart from the 4,3-borocupration, the other possible modes of the borocupration,namely 3,4-,1,2-and 2,1-borocupration,were considered in order to investigate the regioselectivity of the reaction(see Supporting information for other possibilities).As depicted in Fig.3,the computations indeed show that the energies of the other possibilities of the borocupration are much higher than that of the 4,3-borocupration by at least 5 kcal/mol,which is in consistent with the fact that only 4,3-borocyanation products were observed in the experiments [7,11].
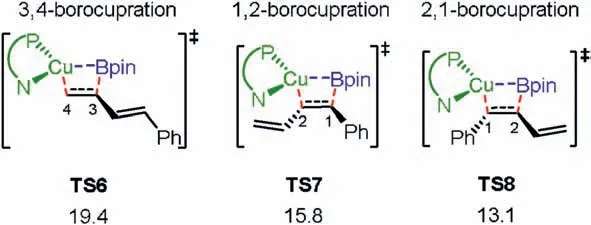
Fig.3.Other possibilities of the borocupration.Energies are given in kcal/mol.
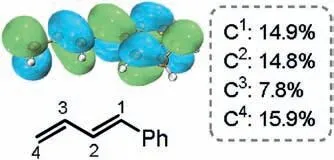
Fig.4.LUMO analysis of 1a.The numbers shown in box are the orbital percentage contributions of the carbon atoms.
Previous studies have demonstrated that the nature of the borocupration is the nucleophilic attack of the boryl group on the alkenes, and the orbital percentage contributions of the carbon atoms in the lowest unoccupied molecular orbital (LUMO) of alkene play an important role in determining the regioselectivity[6a,b].The LUMO analysis of 1a indeed correlates quite well with this finding,showing that the contribution of the C4atom is among the highest (Fig.4).This is due to the presence of the π-electron withdrawing phenyl group at the C1atom makes the C4atom being more electrophilic than the other carbon atoms,which thus results in the addition of the Bpin group to the C4atom being favorable.Further to this,the steric repulsion around the forming C--B bond in the 4,3-borocupration transition state should be smaller than that in the other transition states(see Supporting information for the optimized geometries), which also contributes to the experimentally observed exclusive 4,3-regioselectivity.
To summarize, we have herein presented a mechanistic study on the copper-catalyzed borocyanation of 1-aryl-1,3-butadienes by means of DFTcalculations.The computations show that the overall catalytic cycle consists of five major steps, including (1) the borocupration of the C=C double bond,(2)the η1-allyl isomerization of the allyl copper intermediate, (3) the electrophilic cyanation via the six-membered transition state, (4) the 1,2-elimiantion of the copper tosylamide, and (5) the regeneration of the active catalyst.The borocupration was found to be the rate-and selectivity-determining step of the overall reaction.The presence of the π-electron withdrawing phenyl group at the C1atom makes the C4atom being more electrophilic than the other carbon atoms,which coupled with the steric repulsion around the forming C--B in the borocupration transition states, leading to the experimentally observed exclusive 4,3-borocyantion.The origins of the enantioselectivity are due to the combination of both steric and electronic effects.The steric repulsion between the butadiene moiety and the oxazoline ring of the ligand and the π-π stacking interaction between the phenyl group of the butadiene and the aryl ring of the ligand results in the S-transition state being more favorable than the R-transition state,enabling the experimentally observed good enantioselectivity.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Natural Science Foundation of China(Nos.22073066,21503143 and 21975179)and the Natural Science Foundation of Tianjin (No.16JCQNJC05600).
Appendix A.Supplementary data
Supplementarymaterialrelatedtothisarticlecanbefound,inthe online version,at doi:https://doi.org/10.1016/j.cclet.2020.11.045.
杂志排行

Chinese Chemical Letters的其它文章
- Diverse synthesis of the C ring fragment of bryostatins via Zn/Cu-promoted conjugate addition of α-hydroxy iodide with enone
- Directly conversion the biomass-waste to Si/C composite anode materials for advanced lithium ion batteries
- Recent advances in the improvement of g-C3N4 based photocatalytic materials
- In-situ electro-deposition synthesis of MnOx-NiCo2O4 monolithic catalyst with rich phase interfaces
- Aconapelsulfonines A and B, seco C20-diterpenoid alkaloids deriving via Criegee rearrangements of napelline skeleton from Aconitum carmichaelii
- An unexpected iron(II)-promoted reaction of N-arylprop-2-yn-1-imines with water: Facile assembly of multi-substituted pyrroles