1例少见V型PML-RARA基因的急性早幼粒细胞白血病报告并文献复习*
2021-04-01盛宏霞谢婧胡国亮蓝三春赵志强杨扬孙婷任婧魏然李欲航胡亮钉张斌
盛宏霞,谢婧,胡国亮,蓝三春,赵志强,杨扬,孙婷,任婧,魏然,李欲航,胡亮钉,张斌
(中国人民解放军总医院第五医学中心血液学医学部,北京 100071)
急性早幼粒细胞白血病(acute promyelocytic leukemia,APL)是急性髓细胞白血病(acute myeloid leukemia,AML)的一种特殊类型,FAB协作组定为AML-M3型。APL约占AML的10%,其中90%的APL伴有t(15;17)(q22;q21)遗传学异常,根据15q22断裂位点的不同,可分为3种PML-RARA亚型,分别为长型(L型)、变异型(V型)和短型(S型),且相应的临床特点不同[1]。但也有一些伴或单独拥有其他少见的X-RARA融合基因的情况[2-3],较少见。我院收治1例t(15;17)(q24;q21)的PML-RARA融合基因少见V型的APL病例,报道如下。
1 病历资料
患者,男,43岁。2019年6月末于南京行常规体检,WBC 2.2×109/L。于南京市鼓楼医院就诊,WBC 2.3×109/L,中性粒细胞计数1×109/L,骨髓细胞形态学检查发现骨髓有核细胞增生减低,G∶E=2.2∶1,异常早幼粒细胞占28%,提示不排除APL;免疫分型提示异常细胞在有核细胞中占比约为26.24%,该群细胞表达CD13、CD33、CD64、CD117,不表达CD34、HLA-DR。上述结果提示APL。为进一步诊治于解放军第307医院住院。入院查体:体温36.9 ℃,神志清楚,无贫血貌,全身皮肤黏膜未见皮疹及出血点,肝脾肋下未触及,WBC 2.09×109/L,Hb 126 g/L,PLT 117×109/L。
入院后复查骨髓细胞形态学:骨髓增生减低-活跃,粒系占有核细胞49.5%,其中多颗粒早幼粒细胞占20.5%,见柴捆样Auer小体,见图1。免疫分型提示45.5%(占有核细胞)为可疑幼稚细胞,该群细胞表达CD117;不表达CD34。染色体核型为46,XY,t(15;17)(q24;q21)[10]/46,XY[14](图2)。FISH分析结果提示PML-RARA融合基因阳性细胞占95.4%,见图3。融合基因定性筛查,PML-RARA(L型、S型、V型)阴性;常规PML-RARA(L型、S型、V型)定量检测,PML-RARA/ABL1为0。血液病相关基因检测:未见与目前已经报道的髓系血液疾病相关的致病性突变相关基因。虽然RT-PCR检测PML-RARA融合基因为阴性,结合其他检测结果诊断为APL(低危型)。经维甲酸+亚砷酸双诱导方案治疗后,行骨穿复检。实验室检查:骨髓增生活跃,原幼单核细胞1.5%,未见特异性早幼粒细胞。免疫分型:未见异常表型细胞。FISH分析:PML-RARA融合基因阳性细胞占0%。考虑为完全缓解状态。后患者出院,转回当地医院继续完成后续巩固治疗。2020年4月1日随访患者,仍为完全缓解状态。
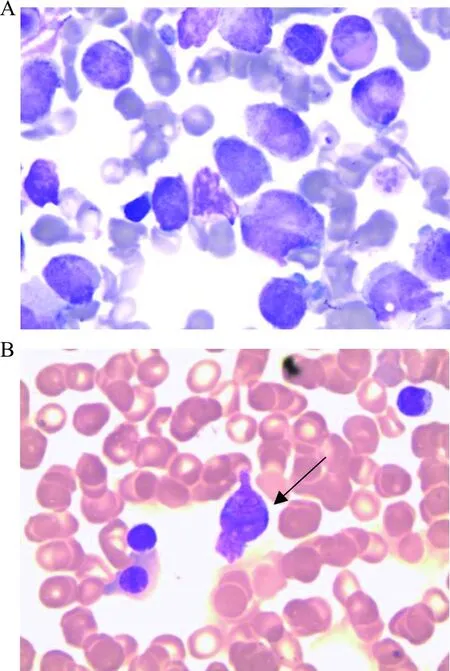
注:A,以多颗粒早幼粒细胞为主;B,箭头示柴捆样Auer小体。
依据染色体核型及FISH结果可见:该患者有PML-RARA的融合基因存在,开始未检出,可能其融合位置发生非常规断裂位点的重排,查询文献[4],重新合成引物进行一代测序,结果示:该标本检测到PML基因的6号外显子发生断裂与RARA基因3号外显子发生融合(见图4),与常见V型不同是,PML6号外显子常规断裂位点缺失25 bp、插入11 bp后与RARA基因的3号外显子连接。且根据测序结果设计定量引物及探针,其序列见表1。将首次入院骨髓样本进行重新定量检测,PML-RARA/ABL1的结果为24.8%。
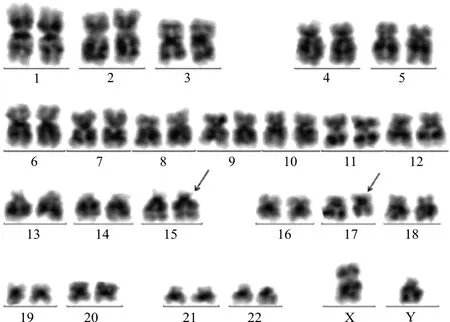
注:G显带示,46,XY,t(15;17)(q24;q21)[10]/46,XY[14]。

注:箭头示PML-RARA阳性细胞。

注:A,该患者骨髓样本部分测序正向图谱,PML基因正常V型断裂位点位于6号外显子,与RARA的3号外显子连接;B,本例患者标本在PML 6号外显子断裂位点与常见不一样,显示缺失25 bp、插入11 bp片段后与RARA基因的3号外显子相连。

表1 该少见V型PML-RARA基因定量检测的引物与探针序列
2 讨论及文献复习
约70%~90%的APL具有特异染色体t(15;17)易位,形成PML-RARA融合基因,这是APL特有的细胞遗传学标志[5]。根据PML的断裂位点不同,PML-RARA基因分3个类型,L型、S型、V型,其中V型只占4%~5%[6],V型断裂位点在PML基因的6号外显子中,且V型患者合并出血的比例低于L型和S型。本文报道1例少见V型PML-RARA的病例。该初治骨髓标本检测发现PML6号外显子常见断裂位点缺失25 bp后插入11 bp的片段,查阅文献且在Cosmic与Clinvar上进行序列比对,V型PML-RARA自身既发生缺失又发生碱基插入的片段的情况未见报道。
本报道患者因行常规体检发现白细胞低而就诊于当地医院,进行检测诊断为APL,予以维甲酸治疗。入我院检查染色体分析结果为46,XY,t(15;17)(q24;q21)[10]/46,XY[14],骨髓细胞形态及免疫表型均支持APL,且FISH检测提示存在PML-RARA融合基因,WBC<10×109/L,综合分析诊断为APL低危型。该病例经过维甲酸+亚砷酸双诱导方案治疗后一直较好,经治疗8个月后仍处于完全缓解状态。
临床上全反式维甲酸诱导分化治疗能使85%左右的APL患者获得缓解,预后较好;极少数无此融合基因者,维甲酸治疗不敏感,预后较差。该患者标本虽然断裂位点与常见V型不同,但由其临床治疗效果可见,该患者对全反式维甲酸诱导分化治疗反应良好。除典型的t(15;17)外,其他变异性的易位也有不少报道。2007年[3]报道1例在幼年型粒单核细胞白血病(juvenile myelomonocytic leukemia,JMML)中由t(4;17)(q12;q21)引起位于4q12的基因FIP1L1和17q21的基因RARA产生的新的FIP1L1/RARA融合基因。也有报道隐匿的PML-RARA基因存在,其断裂位点出现在PML的Exon7b和RARA的Exon3,产生了一个新的PML-RARA转录本[7]。该患者在第2次巩固化疗后获得了分子缓解,并在22个月后仍处于完全缓解状态。Liu等[8]报道了1例IRF2BP2-RARA合并N-RAS突变的APL在经全反式维甲酸和三氧化二砷与柔红霉素结合,治疗后12个月出现了复发,患者对所有其他的化疗都有耐药性。经文献复习提示,对于少见变异性PML-RARA的融合基因,后期治疗效果是否良好可能与其是否合并其他突变有关[8],对于本报道中的少见V型PML-RARA可能需要进行长期的随访及微小残留监测。
实时定量PCR可在99%的典型APL患者中检出PML-RARα融合基因,但仍有1%的APL患者可出现假阴性[9],通过本病例的系统性检测,表明在白血病的诊断检测中合理运用MICM检测有助于患者的诊断及后期的治疗,使患者能得到最大的获益。
