MoS2/SnS2异质结催化性能增强的第一性原理研究
2021-04-01申猛陈昌兆张劲松
申猛,陈昌兆,张劲松
(安徽理工大学力学与光电物理学院,安徽淮南232001)
二维层状材料因其独特的性能和潜在的应用而引起人们的广泛关注,如石墨烯,石墨氮化碳(g-C3N4),过渡金属二卤化合物(如MoS2、SnS2和WS2),由于其独特的几何和电子结构,已被考虑在催化,光电子,储能和传感等领域具有潜在的应用.在这些二维结构中,强平面耦合和弱范德华相互作用促进了新功能和器件的实现,同时具有良好的光催化和电催化活性[1].与二维过渡金属氧化物相比,二维金属硫化物具有更好的优点,如带隙窄(低至1.8 eV),太阳能利用效率高,载流子迁移率高,这都有利于载流子的传输,从而具有更好的催化性能.过渡金属硫化物中的MoS2具有独特的二维层状结构,具有良好的热稳定性和化学稳定性,在储氢和催化等领域具有重要的潜在价值[2-3].二硫化钼固体是一种间接带隙半导体材料,带隙的宽度为1.29 eV[4].单层MoS2是一种直接带隙半导体材料,带隙宽度为1.8 eV[5],是一种新型的二维分层化合物,属于六方体系,MoS2由于具有良好的光学性能和催化性能而被广泛的研究.在单层MoS2晶体结构中,Mo原子与S原子之间以及Mo原子与Mo原子之间形成共价键,是一种“三明治”结构,层间的相互作用受范德华力的约束[6-7].但是它的电子-空穴复合效率高,进而导致光催化性能较低.因此,有必要通过改进技术来抑制MoS2材料中电子-空穴的复合,进而提高光催化性能.
在材料性能改进方面许多学者也进行过深入研究.其中最常用的方法是掺杂,通过控制掺杂的原子以及掺杂原子比例进而调节材料的带隙,从而改变材料的性能[8-9].但是单原子掺杂存在一定的缺陷,比如热稳定性差,电子-空穴复合不能得到有效的抑制等.在此基础上开展了共掺杂方法研究,例如,金属与非金属之间的共同作用等研究.异质结能有效分离光生电子和空穴,进而提高材料的催化性能.在构建异质结研究方面,MABIALA-POATY等学者也做了大量的工作[10-11].构建的异质结除了具有独特的性能外,还会扩大材料的应用范围.二维过渡金属硫化物,SnS2是一种潜在的半导体光催化剂,具有六边形晶体结构,其沿c轴的晶格参数较大(0.59 nm)[12-13].通过调整合成参数,可以很容易地沿着(001)方向分离SnS2层,得到超薄的六边形纳米片[14].超薄的六方SnS2纳米片具有较大的比表面积,其二维表面适合于加载其他半导体以获得良好接触的异质结,进而提高载流子的传输.由于带隙较窄,二维单层MoS2纳米片在构建异质结提高光催化活性方面具有很大的优势.
通过查阅相关文献可知,有关MoS2/SnS2异质结理论的相关文献较少,基于以上思路,在理论上能够通过构建MoS2/SnS2异质结来提高复合载体的输运性能和催化性能.该研究采用密度泛函理论的方法系统地计算了单层MoS2,单层SnS2和MoS2/SnS2异质结的几何结构和电子结构,并对上述理论结果进行了分析.同时,还计算了MoS2/SnS2异质结的差分电荷密度和平面平均差分电荷密度,对体系的电荷转移情况进行了分析.以期为MoS2/SnS2异质结研究提供理论指导.
1 模型搭建与计算方法
研究者所有的理论计算都是基于密度泛函理论的第一性原理VASP(Vienna ab-initio Simulation Package)[15-17],该原理使用的是广义梯度近似Perdew-Burke-Ernzerhof(PBE)[18-19].计算原子的价电子构型分别为Mo:4d55s1,Sn:4d105s25p2,S:3s23p4.平面波截断能为500 eV,布里渊区K值分别为5×5×1,6×6×2和9×9×1来计算相应的单层MoS2,单层SnS2和MoS2/SnS2异质结的几何结构和电子性质.每个原子的能量和力的收敛精度分别为1×10-5eV和0.01 eV/10-10m.为了得到更精确的优化结果,采用DFT-D2方法[18]修正了范德瓦尔斯(vdW)相互作用力,计算了优化后体系的几何结构,电子结构和差分电荷电荷密度等参数.
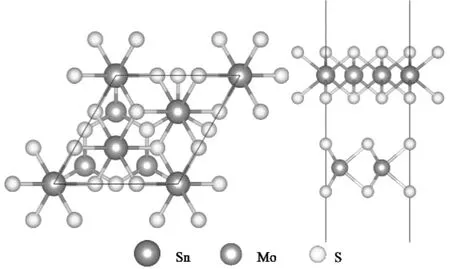
图1 MoS2/SnS2异质结俯视图和正视图Fig.1 MoS2/SnS2 heterojunction view and elevation view
MoS2/SnS2异质结的建立分别由MoS2(001)表面和SnS2(001)表面的超胞体系构成,理论计算了单层MoS2、单层SnS2和MoS2/SnS2异质结的几何和电子结构.通过计算功函数和差电荷密度,进一步分析异质结的电荷分布,揭示了MoS2/SnS2异质结的界面性质,光诱导载流子转移机理和光催化性质之间的内在关系.构建的异质结晶格失配率约为0.35%,为使MoS2/SnS2异质结相邻体系间的相互作用最小化,采用15×10-10m真空能,优化后的MoS2/SnS2异质结的结构模型如图1所示.
2 结果与讨论
2.1 晶体结构
在构建MoS2/SnS2异质结之前,首先对单层MoS2和单层SnS2进行几何优化,使其达到能量最低值.对于单层MoS2,结构优化后的晶格参数为a=b=3.19×10-10m,与其他理论结果a=b=3.18×10-10m[20-22]吻合较好.对于单层SnS2,结构优化后的晶格参数为a=b=3.69×10-10m,c=5.98×10-10m,与其他理论结果a=b=3.64×10-10m,c=5.88×10-10m[23-24]吻合较好,表明构建异质结的科学性.
2.2 能带结构与态密度分析
为了准确分析MoS2/SnS2异质结体系的电子结构特性,理论计算了单层MoS2,单层SnS2和MoS2/SnS2异质结的能带结构和态密度,进而研究MoS2/SnS2异质结的光催化性能,其能带结构和态密度如图2所示.
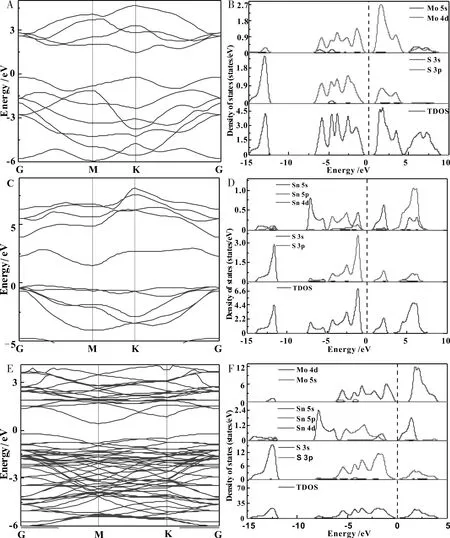
图2 A,B单层MoS2;C,D单层SnS2;E,FMoS2/SnS2异质结的能带结构和态密度Fig.2 A,BSingle layer MoS2;C,DSingle-layer SnS2;E,Fenergy band structure and state density of MoS2/SnS2 heterojunction
如图可知单层MoS2是一种直接带隙半导体,其中VBM和CBM均位于K点处,计算得到的带隙为1.67 eV,与之前[25]的计算结果吻合较好.由图2-C可知,单层SnS2是一种间接带隙半导体,其中VBM位于G点,而CBM位于G和M点之间,计算得出的带隙为1.73 eV,与其他计算结果[26]吻合较好.从图2-E可以看出,MoS2/SnS2异质结为间接带隙半导体,带隙大小为0.66 eV.MoS2/SnS2异质结的态密度和分波态密度如图2-F所示,MoS2/SnS2异质结的CBM被Sn和S轨道占据,而MoS2/SnS2异质结的VBM被Mo轨道占据.因此,MoS2/SnS2异质结的CBM和VBM属于不同的组分,为MoS2和SnS2共同贡献,态密度的分析与能带结构的结果相一致.
与单层MoS2和单层SnS2的能带结构相比,MoS2/SnS2异质结的能带结构并不是单层MoS2和单层SnS2能带结构的简单叠加,其能带结构受MoS2/SnS2界面间范德华力作用的影响,MoS2/SnS2异质结的带分布在禁带附近交错,在导带中,SnS2的电子轨道位于导带的较低能级,而MoS2的电子轨道位于导带中的较高能级.在价带中,SnS2的电子轨道位于价带的较低能级,MoS2的电子轨道位于价带的较高能级.在MoS2/SnS2异质结中,在导带中主要是由SnS2占据,在价带中主要是由MoS2占据.上面的理论结果分析表明,MoS2/SnS2异质结的带隙(0.66 eV)明显低于单层MoS2的带隙(1.67 eV)和单层SnS2的带隙(1.53 eV),构建的异质结使电子很容易从价带跃迁到导带,从而提高对可见光的吸收能力.从态密度中可以看出,MoS2/SnS2异质结体系的局域化强度增强,能带更致密,带间波动更加平缓.异质结的构建减小了能带带隙,降低了电子跃迁所需的能量,异质结的交错能带可以促进光生载流子的分离,从而提高了体系的光催化活性.
2.3 功函数与差分电荷密度
表面功函数是研究界面电荷转移和半导体能带对准的一个重要参数.我们计算了单层MoS2和单层SnS2的功函数见图3.
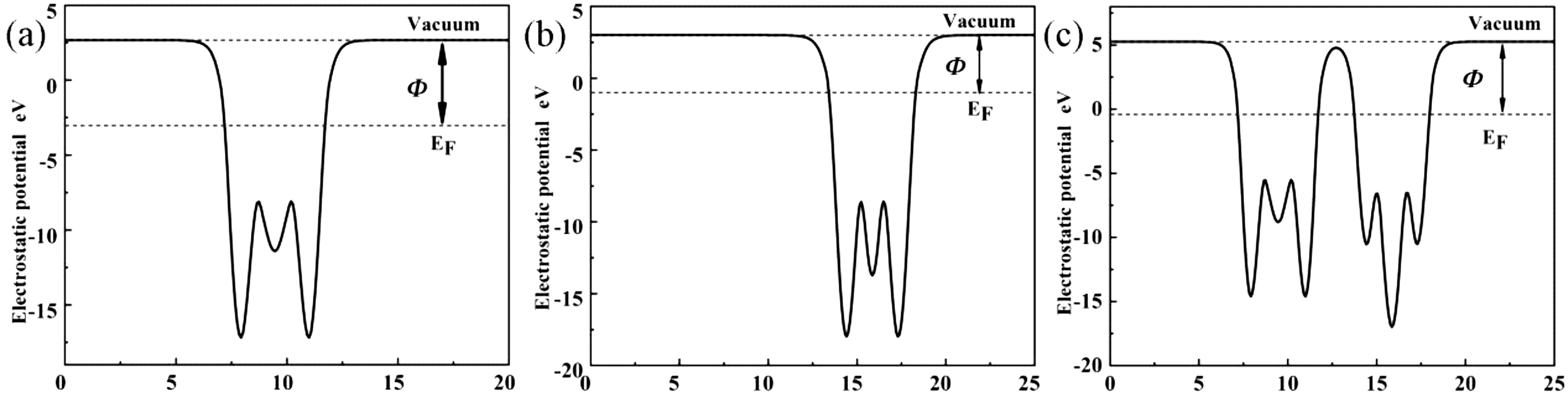
图3 A单层MoS2,B单层SnS2,C MoS2/SnS2异质结的功函数Fig.3 A monolayer MoS2,Bmonolayer SnS2,Cwork function of MoS2/SnS2 heterojunction
根据以下公式[26]

式(1)中:Φ代表功函数,EVAC代表的真空能级,EF代表是费米能级.理论计算了单层MoS2,单层SnS2和MoS2/SnS2异质结的功函数分别为5.71 eV,4.07 eV和5.67 eV,与之前的计算结果一致[27-29].通过比较单层的MoS2和单层SnS2的功函数可以看出,单层SnS2纳米片的功函数低于单层MoS2纳米片的功函数.当单层MoS2与单层SnS2相互作用形成MoS2/SnS2异质结时,电子会从SnS2表面流向MoS2表面,直到达到相同的费米能级为止.MoS2和SnS2的表面由于电子的转移而带电,MoS2表面聚集了负电荷,而SnS2表面聚集了正电荷,在界面会形成一个内电场,电场的方向从MoS2表面指向SnS2表面.
为了进一步分析MoS2/SnS2异质结界面间的电荷转移和分离,通过计算体系的差分电荷密度见图4.
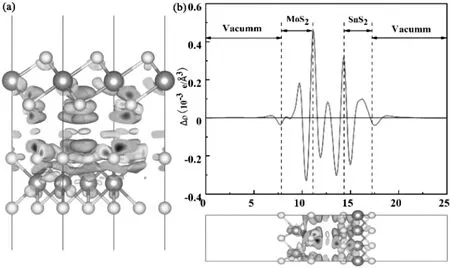
图4 MoS2/SnS2异质结的电荷密度Fig.4 The chargedensity of MoS2/SnS2 heterojunctions
图4-A,4-B分别为MoS2/SnS2异质结的差分电荷密度,平面平均差分电荷密度.计算公式如下[30]

式(2)中:ρMoS2/SnS2,ρMoS2和ρSnS2分别代表MoS2/SnS2异质结,单层MoS2和单层SnS2的电荷密度.计算得到的MoS2/SnS2异质结的电荷密度差如图4-A所示,黄色区域表示电子积累,青色区域表示电子消耗.电荷重分布主要发生在MoS2界面附近,而SnS2内部的电荷密度变化很小.为了进一步探究MoS2/SnS2的电子转移,理论计算了异质结沿Z方向的平面平均差分电荷密度,如图4-B所示.可以从MoS2/SnS2异质结界面处电荷密度的变化看出,电子主要通过界面从SnS2转移到MoS2,而电子主要停留在MoS2侧,空穴则停留在SnS2侧,这与上述功函数的分析结果是一致的.在它们的界面处形成一个内置电场,电场方向从SnS2的表面到MoS2的表面.基于以上结果分析,异质结的构建可以有效提高复合体系的电荷转移能力,抑制光生电子和空穴的复合,从而提高材料的光催化性能.
2.4 异质结光催化机理分析
结合单层MoS2,单层SnS2的带隙以及它们功函数的计算,对MoS2/SnS2异质结的光催化机理进行了探究,异质结的光催化机理图见图5.
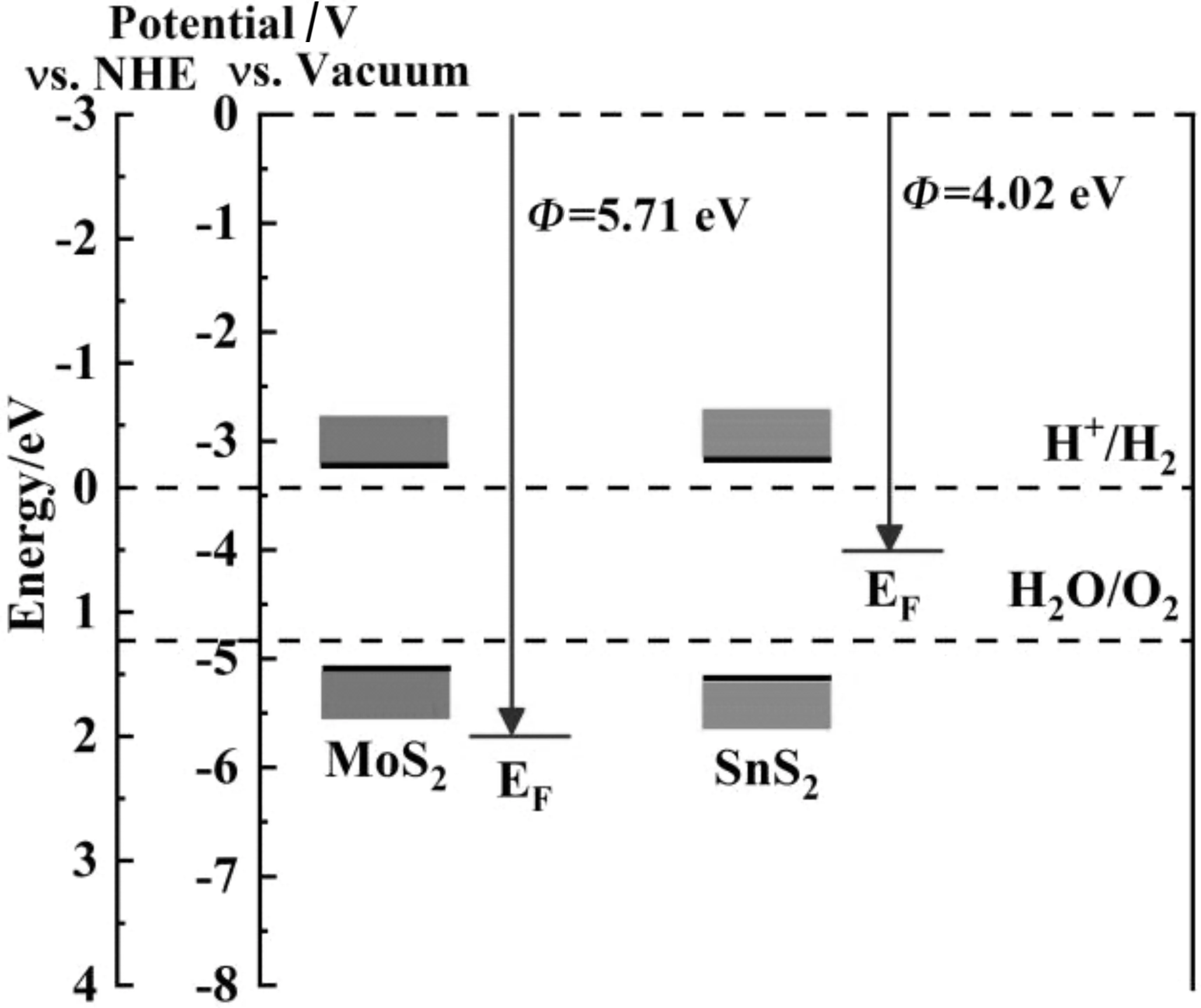
图5 MoS2/SnS2异质结构光催化机理Fig.5 Photocatalytic mechanismof MoS2/SnS2 heterostructure
从图5中可以看出MoS2和SnS2的能带分布.在光照下,当MoS2与SnS2相接触,SnS2中的电子通过界面向MoS2转移,并形成内置电场,内置电场的存在不利于SnS2中光生空穴转移到MoS2中,然而有利于SnS2的光激发电子向MoS2跃迁.这样MoS2积累了更多的光生电子,SnS2积累了更多的光生空穴,这样光激发电子和空穴在空间上被有效地分离,诱导了更多的光激发电子和空穴参与表面的光催化反应,从而提高了光催化效率.
3 结论
本文采用第一性原理平面波超软赝势分析方法,通过构建MoS2/SnS2异质结构,计算并分析了其能带结构,态密度,功函数和差分电荷密度等性能.理论计算得到MoS2/SnS2异质结体系的带隙为0.66 eV,更小的带隙则有利于可见光的捕获.功函数的结果分析表明,电子会从SnS2的表面流向MoS2的表面.差分电荷密度和平面平均差分电荷密度的结果分析表明,在光照下,内部电场促进了MoS2/SnS2异质结光生电子和空穴的分离以及抑制了光生电子和空穴的复合,体系得到了更优异的光催化性能,同时为实验上合成MoS2/SnS2提供了一定的理论指导.
