SmProt:A Reliable Repository with Comprehensive Annotation of Small Proteins Identified from Ribosome Profiling
2021-03-30YanyanLiHonghongZhouXiaominChenYuZhengQuanKangDiHaoLiliZhangTingruiSongHuaxiaLuoYajingHaoRunshengChenPengZhangShunminHe
Yanyan Li ,Honghong Zhou ,Xiaomin Chen ,Yu Zheng ,Quan Kang ,Di Hao ,Lili Zhang,3,Tingrui Song,Huaxia Luo,Yajing Hao,Runsheng Chen,3,5,*,Peng Zhang,*,Shunmin He,*
1College of Life Sciences,University of Chinese Academy of Sciences,Beijing 100049,China
2Key Laboratory of RNA Biology,Center for Big Data Research in Health,Institute of Biophysics,Chinese Academy of Sciences,Beijing 100101,China
3University of Chinese Academy of Sciences,Beijing 100049,China
4Department of Cellular and Molecular Medicine,University of California,San Diego,La Jolla,CA 92093,USA
5Guangdong Geneway Decoding Bio-Tech Co.Ltd,Foshan 528316,China
Abstract Small proteins specifically refer to proteins consisting of less than 100 amino acids translated from small open reading frames(sORFs),which were usually missed in previous genome annotation.The significance of small proteins has been revealed in current years,along with the discovery of their diverse functions.However,systematic annotation of small proteins is still insufficient.SmProt was specially developed to provide valuable information on small proteins for scientific community.Here we present the update of SmProt,which emphasizes reliability of translated sORFs,genetic variants in translated sORFs,disease-specific sORF translation events or sequences,and remarkably increased data volume.More components such as non-ATG translation initiation,function,and new sources are also included.SmProt incorporated 638,958 unique small proteins curated from 3,165,229 primary records,which were computationally predicted from 419 ribosome profiling (Ribo-seq) datasets or collected from literature and other sources from 370 cell lines or tissues in 8 species (Homo sapiens, Mus musculus, Rattus norvegicus, Drosophila melanogaster, Danio rerio, Saccharomyces cerevisiae, Caenorhabditis elegans,and Escherichia coli).In addition,small protein families identified from human microbiomes were also collected.All datasets in SmProt are free to access,and available for browse,search,and bulk downloads at http://bigdata.ibp.ac.cn/SmProt/.
KEYWORDS Ribosome profiling;Small open reading frame;Upstream open reading frame;Variants;Disease
Introduction
Genome annotation is fundamental to life science.In recent years,it has been found that small open reading frames(sORFs) widely exist in genomes of many organisms including humans [1] and human microbiomes [2],and some are able to be translated into small proteins [3-5].Small proteins are proteins with less than 100 amino acids,which may be derived from untranslated regions(UTRs)of mRNAs[6]or non-coding RNAs(ncRNAs)[7,8]including primary microRNAs (pri-miRNAs) [9,10],long ncRNAs(lncRNAs)[11],and circular RNAs(circRNAs)[12].Small proteins were usually missed in previous coding sequence annotation,while their significance has been revealed in current years for diverse functions[13],such as embryonic development [14,15],cell apoptosis [16],muscle contraction [17],and antimicrobial activity [18].Some small proteins play roles in multiple diseases [19,20] including tumors[9,11,12].Despite the abundance of sORFs in genome,the number of well-studied small proteins is very limited.Annotation of numerous small proteins will contribute to studies on various physiological and pathological processes.
Identification of small proteins at proteomic level is challenging.Mass spectrometry (MS) can provide direct evidence of small proteins,but it relies much on the coverage of existing libraries,which mainly focus on large proteins rather than small proteins.Protease cleavage sites are lacking in small proteins due to the limited length.Besides,small proteins are usually of low abundance,and tend to be filtered out during enrichment process[21].Ribosome profiling (also named as ribosomal footprinting or Riboseq) provides a more sensitive way for global detection of translation events based on the deep sequencing of ribosome-protected mRNA fragments (RPFs) [22,23],which allows for identifying the location of translated ORFs and translation initiation sites (TISs),the distribution of ribosomes on mRNA,and the speed of translating ribosomes [24].Reference libraries for MS can also be constructed with Ribo-seq data.The regular Ribo-seq (rRiboseq)utilizes cycloheximide(CHX)[25],a drug bound at the ribosome E-site[26],as a translation elongation inhibitor to freeze the translating ribosomes.Translation is principally regulated at the initiation stage.Translation initiation sequencing(TI-seq)is a variation of rRibo-seq technique that uses different translation inhibitors,usually lactomidomycin(LTM) [25] or harringtonine (HARR) [27],which can induce ribosome stasis at TISs.TI-seq enables the global mapping of TISs,and is more accurate in prediction of non-ATG start codons.Many sORFs are proved to use nonclassical ATG start codons[28],which is also an important mechanism for generating protein isoforms [29,30].rRiboseq data usually show clear triplet periodicity[26].Different computational analysis strategies[31-38]have been developed to identify translated sequences using Ribo-seq data.
Emerging evidence shows that many upstream ORFs(uORFs)act incisto regulate the translation of downstream ORFs by leaky scanning [39],reinitiation [40],and ribosome stalling [41].Recently,variants creating new upstream start codons or disrupting stop sites of existing uORFs (uORF-perturbing) are found to be under strong negative selection [42].uORF-perturbing variants have been demonstrated as an under-recognized functional class that contribute to human disease.
Given the great importance of small proteins,in-depth investigations of small proteins across various species are in need.SmProt is dedicated to integrating knowledge of small proteins translated from various sources,especially for those from UTRs and ncRNAs.The annotation information and functional sections in the current release are much richer than those in the initial release [43],and the data volume and reliability are also greatly improved.
Data collection and processing
Data sources
rRibo-seq and TI-seq datasets derived from diverse tissues/cell lines were collected from GEO [44] and European Nucleotide Archive (ENA) [45] databases.The latest reference genomes and gene annotations were download from Ensembl [46],GENCODE [47],and NCBI-Genome database.Whole-genome sequencing (WGS) variants were collected from various websites.The construction pipeline of SmProt is summarized inFigure 1.

Figure 1 Construction of SmProt
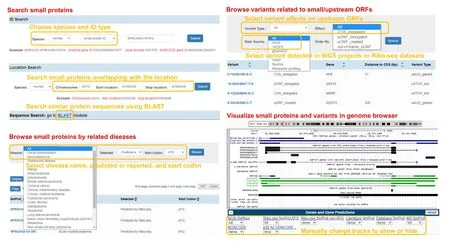
Figure 2 Usage of SmProt
Ribo-seq data processing
The fastq files of 547 Ribo-seq datasets were downloaded from GEO and ENA databases.Each dataset was checked manually to confirm the sequencing adapters.The adapters were removed using cutadapt 1.18[48]and only reads with 25-35 bp in length were retained.Then the sequences were mapped to the latest genome using STAR 2.5.2a[49]using EndToEnd mode with allowance of up to 2 mismatches.
Ribo-seq quality and P-site offsets were assessed by Ribo-TISH [34] quality module.For TI-seq data,more attention was put on TIS quality (-t).Manual checks were then carried out to verify offset values and eliminate datasets without obvious triplet periodicity.After the quality control,419 Ribo-seq datasets (Table S1) were retained.
Translated ORFs were predicted by Ribo-TISH predict module.Biological and technical duplication data under the same treatment in one dataset were merged.Minimum amino acid length of candidate ORFs was set to 5.Considering both ATG and near-cognate start codons(with one base different from ATG),rRibo-seq datasets using only CHX without matched TI-seq data were analyzed twice.One is prediction of ORFs with canonical ATG start codon,the other is prediction of ORFs with near-cognate start codons (--alt).Preferring data evidence instead of prior assumption in our database,only the best frame test results from multiple candidate start codons in the same ORF were reported(--framebest).For datasets containing TI-seq data,alternative start codons were included (--alt),and different parameters were set for LTM-based TI-seq and HARR-based TI-seq(--harr).
sORFs with less than 100 amino acids were filtered from the prediction results above.Furthermore,we removed some prediction results that may be supported by RPFs from other classic proteins with more than 100 amino acids.These include ORFs marked asknown(i.e.,TIS annotated in another transcript),CDSFrameOverlap(i.e.,ORF overlapping with annotated CDS in another transcript in the same reading frame),andTruncated(i.e.,ORF as a part of annotated CDS in the same transcript) without translation initiation evidence (i.e.,no significant results identified from paired TI-seq datasets).
In-frame reads of sORFs were counted and normalized by library sequencing depth(in-frame total reads count)and sORF length,a similar method with reads per kilobase per million mapped reads (RPKM) in RNA-seq but using ribosome profiling data that represents the translation levels.
Finally,3,060,793 records (i.e.,unmerged primary results from all datasets and tissues) were retained.Results with the identical genome loci in one species were merged as the same small protein,generating 577,206 unique IDs,while information derived from multiple datasets were retained,a similar integration method as for piRBase [50].
Variants from ribosome profiling data
We performed germline variants detection on 96 human ribosome profiling datasets,referring to the workflow for processing RNA data for germline short variant discovery with GATK v4.1.8[51-54].Duplicate reads were identified using MarkDuplicates tool after alignment,then reads with unidentified nucleotide (N) in Cigar were split using SplitNCigarReads tool.Base quality score recalibration was carried out based on true sites in training sets using BaseRecalibrator tool and applied using ApplyBQSR tool.Variants were called individually in each sample using the HaplotypeCaller tool.Variants with QualByDepth(QD)<2 were removed using VariantFiltration tool.Germline single nucleotide variants(SNVs)were linked to small proteins in SmProt according to genomic positions.
Variants from WGS data
Variants from 1KGP3 [55],GAsP [56],TOPMed [57],gnomAD3 [42,58],and NyuWa [59] were collected.VCF files were lifted over from old genome version to GRCh38 using LiftoverVcf tool of GATK with allowance to recover swapped reference and alternative alleles.
Variants in 5′ UTRs were evaluated for their effects on uORFs using VEP[60]with plugin UTRannotator[42,61],and classified by their functional consequences.Translation evidence of uORFs was based on small proteins recorded in SmProt.
Disease-specific small proteins
Small proteins identified only from diseased cell lines/tissues but not from corresponding normal cell lines/tissues were predicted as disease-specific translation events.If there were matched data of normal and diseased groups in the same dataset,small proteins derived uniquely from diseased group were screened as disease-specific ones.If there was no matched control group in the same dataset,the same type of healthy tissue/cell line in other datasets were used as control.If there was no matched same tissue/cell line,all data from diverse normal tissues/cell lines were merged for comparisons (Table S2),and small proteins identified only from the diseased cell lines/tissues were predicted as tissue-specific.Disease-specific or tissue-specific translation events require RiboPvalue in disease groups lower than 0.01 while similar proteins with different TISs at the same loci in control group not detected(RiboPvalue higher than 0.05).
SNVs in diseased cell lines/tissues derived from ribosome profiling data and located within the genomic region of small proteins were regarded as diseased variant sets.SNVs detected only in diseased variant sets but not in normal sets were predicted as disease-specific SNVs.SNVs in corresponding normal cell lines/tissues (Table S2) derived from ribosome profiling data were combined with all variants derived from multiple WGS projects,as control variant sets for comparison.
Function domain prediction
Besides function of small proteins collected from literature mining,we used InterProScan [62] to predict function domain of small proteins,which focuses on combination of protein family membership and the functional domains/sites,and has been extensively used by genome sequencing projects and the UniProt Knowledgebase [63].Default thresholds and additional parameters-goterms -pawere adopted for gene oncology and pathway annotations.
PhyloCSF calculation
Pre-calculated BigWig data of PhyloCSF[64]scores at each base across the whole genome were downloaded from Broad Institute (https://data.broadinstitute.org/compbio1/PhyloCSFtracks/),and the score for genomic region of each small protein was extracted with our script using pyBigWig(https://github.com/deeptools/pyBigWig).
Database implementation
Database website was organized with HTML (https://html.spec.whatwg.org/),JavaScript (https://www.javascript.com/),PHP (https://www.php.net/),and MYSQL (https://www.mysql.com/).UCSC Genome Browser(http://genome.ucsc.edu/)was used to visualize the small proteins and variants.NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi)was used for sequence similarity searches.
Database content and usage
Overview
SmProt was constructed by pipeline described in Figure 1.Multiple ways were provided to search,browse,visualize,and study small proteins (Figure 2).Small proteins were found mainly from rRibo-seq and TI-seq data.All information for small proteins from different data sources and datasets were integrated.General information for small proteins was provided such as sequence,mass,location,blocks,tissue or cell line,predicted functions,conservation,and multiple IDs including small protein ID,Ensembl ID,and NONCODE[65]ID.Translation level(in frame counts and Ribo RPKM) of small proteins identified from each dataset and record was provided.Details for their related variants and diseases were also provided (Figure 3).SmProt now has 638,958 unique small proteins and 3,165,229 small protein records in total(Table 1;Table S3).
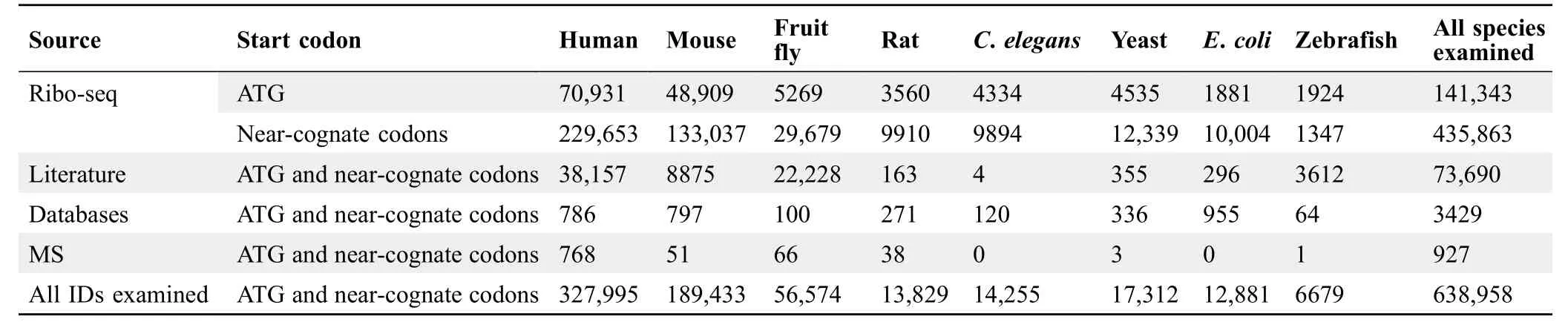
Table 1 Statistics of unique small proteins in SmProt

Figure 3 Contents of SmProt
Reliability of small proteins
SmProt emphasizes reliability of small proteins,which is ensured mainly by the significance of 3-nt periodicity in RPF P-site profile.
Firstly,we constructed new pipeline based on independently published toolkit Ribo-TISH[34],which allows for accurate detection of ORFs and TISs using rRibo-seq and TI-seq.Ribo-TISH uses rank sum test to detect 3-nt periodicity,and uses negative binomial test to detect TISs,which outperforms other established methods in prediction accuracy.
Secondly,in addition to the quality control based on Ribo-TISH quality module,manual checks were also carried out to ensure clear triplet periodicity and unambiguous offset of Ribo-seq data,which further eliminates noises.
Thirdly,we provided several evaluations as supporting evidence.These include 1)Pvalues of small proteins called from multiple ribosome profiling datasets,which indicate the confidence in different samples and conditions;2)PhyloCSF conservation of genomic regions,which reflects coding potential;and 3) peptide evidence derived from mass spectrum data.All these lines of evidences are exhibited in the small protein page.Moreover,a set with evidence of both translation events and protein fragments is provided on download page.
In addition,information of small protein derived from multiple sources is also integrated in small protein information page.
Variants related to small proteins
In total,25,475 variants located on translated sORFs were provided,which are on display in the related small protein page.Given that uORF-perturbing variants are likely to impact translation of downstream proteins [42],variants from multiple WGS projects and ribosome profiling data were evaluated for their effects on uORFs.These include creating a new start codon ATG,removing an existing start codon ATG,creating a new stop codon within an existing uORF,removing the stop codon of an existing uORF,and creating a frameshift mutation in an existing uORF,which can be found at variants page.
Disease-specific small proteins
Disease-specific small proteins are potential candidates of molecular markers or targets for diagnosis and treatment.Disease-specific translation events as well as diseasespecific SNVs of small proteins in 16 types of diseases were identified (see "Data collection and processing" section)(Table S4).Besides,small proteins that have been verified experimentally in certain diseases were also documented through literature mining.
Human microbiome small proteins
Over 4000 conserved small protein families identified from human microbiomes were collected [2].A new sectionHumanMicroBiowas created to integrate and display selected information of these small protein families.
Other sources
We use a set of keywords(File S1)to search articles about small proteins in PubMed database.High-confidence small proteins in CCDS [66] and Swiss-Prot [67] were also integrated.Literature mining is processed in stages,and the newly-published data from other sources will be released continuously after completion of manual review and curation.
Function domain prediction
For successfully predicted functions of small proteins derived from ribosome profiling and literature mining,SmProt provides graph for visualization and prediction details including Gene Oncology (GO) and pathway annotations.Users can choosepredicted functionsonBrowsepage to filter the results with function domain prediction.
Inner BLAST
The abundant small proteins across multiple species allow for sequence similarity searches at both nucleotide and protein levels.Users can search for sequences of interests using BLASTp and BLASTx (NCBI BLAST 2.2.24 release)online.
Visualization using UCSC Genome Browser
SmProt incorporates UCSC Genome Browser [68] to visualize all the information including genomic loci of small proteins,as well as variants from ribosome profiling data and multiple WGS projects related to small proteins,MS data,and gene annotations.The latest genome versions including hg38,mm10,rn6,dm6,ce11,sacCer3,and danRer11 were provided.
Comparison with other databases
SmProt currently includes 419 Ribo-seq datasets derived from 116 cell lines/tissues,compared to 60 datasets derived from 37 cell lines/tissues in the initial version.The number of small protein records identified from ribosome profiling in the current release is 60 times that of the initial release(3 millionvs.0.05 million).The current release of SmProt combined a large amount of duplicate records in the initial release[43],and Ribo-seq analysis pipeline was optimized to ensure the reliability of our results.Variants in translated sORFs identified from Ribo-seq data as well as uORFperturbing variants identified from WGS projects were provided.Disease-specific small proteins may provide new perspectives for clinical studies.
Currently,there are a few databases for small proteins such as ARA-PEPs [69],PsORF [70],and sORFs.org [71].ARA-PEPs and PsORF only harbor small proteins in plants.sORFs.org developed simple inner TIS-calling algorithm not based on triplet periodicity,which should be the most important feature of Ribo-seq.SmProt emphasizes high confidence using our Ribo-TISH pipeline that is more accurate than previous methods.In total,419 Ribo-seq datasets have been analyzed in SmProt,while there were only 78 Ribo-seq datasets in sORFs.org.Moreover,SmProt pays special attention to function,variants,and related diseases of small proteins.Furthermore,WGS data resources are also integrated in SmProt,which are not covered in other databases.
Other proteomic databases such as UniProt,neXtProt[72],andOpenProt [73] are not specifically designed for small proteins.neXtProt only harbors proteins of humans while SmProt harbors small proteins of 8 species.Simialr to SmProt,OpenProt also used ribosome profiling and mass spectrum to predict proteins including some small proteins longer than 30 amino acids.Nonetheless,SmProt has analyzed many more ribosome profiling datasets (419),which are about 5 times that in OpenProt (87),and provides information for small proteins longer than 5 amino acids.
Conclusion
In brief,SmProt integrates small proteins from large amount of ribosome profiling data,and provides more abundant details.We strongly believe that SmProt will provide valuable and accurate information on small proteins for scientific community.Moreover,SmProt provides a new resource for users interested in functional and mechanistic studies,and a reference for construction of MS libraries of small proteins.
Data availability
SmProt is publicly available at http://bigdata.ibp.ac.cn/SmProt/.
CRediT author statement
Yanyan Li:Conceptualization,Methodology,Investigation,Formal analysis,Data curation,Writing -original draft,Software,Visualization.Honghong Zhou:Investigation,Data curation,Funding acquisition.Xiaomin Chen:Investigation,Data curation.Yu Zheng:Data curation,Software,Visualization.Quan Kang:Software,Visualization.Di Hao:Data curation,Software.Lili Zhang:Visualization.Tingrui Song:Visualization.Huaxia Luo:Writing -review&editing.Yajing Hao:Writing-review&editing.Runsheng Chen:Resources,Supervision,Funding acquisition.Peng Zhang:Conceptualization,Methodology,Investigation,Software,Writing -review &editing,Visualization,Project administration,Funding acquisition.Shunmin He:Conceptualization,Methodology,Resources,Investigation,Writing -review &editing,Supervision,Funding acquisition.All authors have read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the National Key R&D Program of China (Grant No.2016YFC0901702);National Natural Science Foundation of China (Grant Nos.81902519,91940306,31871294,31701117,and 31970647);the National Key R&D Program of China(Grant Nos.2017YFC0907503,2016YFC0901002,and 2018YFA0106901);the Strategic Priority Research Program of Chinese Academy of Sciences (Grant No.XDB38040300);the 13th Five-year Informatization Plan of Chinese Academy of Sciences(Grant No.XXH13505-05);Special Investigation on Science and Technology Basic Resources,Ministry of Science and Technology,China(Grant No.2019FY100102);and the National Genomics Data Center,China.We thank Center for Big Data Research in Health(http://bigdata.ibp.ac.cn/),Institute of Biophysics,Chinese Academy of Sciences,for supporting data analysis and computing resource.We thank Prof.Yiwen Chen from The University of Texas MD Anderson Cancer Center,USA for thoughtful discussions and valuable comments on the database.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2021.09.002.
ORCID
0000-0001-5256-6696 (Yanyan Li)
0000-0001-7409-3092 (Honghong Zhou)
0000-0002-0633-2984 (Xiaomin Chen)
0000-0003-4936-8407 (Yu Zheng)
0000-0001-6790-5259 (Quan Kang)
0000-0003-0082-0730 (Di Hao)
0000-0002-3601-0150 (Lili Zhang)
0000-0003-2967-7704 (Tingrui Song)
0000-0001-9944-0345 (Huaxia Luo)
0000-0003-1384-4176 (Yajing Hao)
0000-0001-6049-8347 (Runsheng Chen)
0000-0001-9303-1639 (Peng Zhang)
0000-0002-7294-0865 (Shunmin He)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- SPA:A Quantitation Strategy for MS Data in Patient-derived Xenograft Models
- RePhine:An Integrative Method for Identification of Drug Response-related Transcriptional Regulators
- NOGEA:A Network-oriented Gene Entropy Approach for Dissecting Disease Comorbidity and Drug Repositioning
- DeepCAPE:A Deep Convolutional Neural Network for the Accurate Prediction of Enhancers
- The Genome Sequence Archive Family:Toward Explosive Data Growth and Diverse Data Types
- Genome Warehouse:A Public Repository Housing Genomescale Data