SPA:A Quantitation Strategy for MS Data in Patient-derived Xenograft Models
2021-03-30XiChengLiliQianBoWangMinjiaTanJingLi
Xi Cheng ,Lili Qian ,Bo Wang ,Minjia Tan ,Jing Li,*
1Department of Bioinformatics and Biostatistics,School of Life Sciences and Biotechnology,Shanghai Jiao Tong University,Shanghai 200240,China
2The Chemical Proteomics Center and State Key Laboratory of Drug Research,Shanghai Institute of Materia Medica,Chinese Academy of Sciences,Shanghai 201203,China
3University of Chinese Academy of Sciences,Beijing 100049,China
Abstract With the development of mass spectrometry (MS)-based proteomics technologies,patient-derived xenograft(PDX),which is generated from the primary tumor of a patient,is widely used for the proteome-wide analysis of cancer mechanism and biomarker identification of a drug.However,the proteomics data interpretation is still challenging due to complex data deconvolution from the PDX sample that is a cross-species mixture of human cancerous tissues and immunodeficient mouse tissues.In this study,by using the lab-assembled mixture of human and mouse cells with different mixing ratios as a benchmark,we developed and evaluated a new method,SPA (shared peptide allocation),for protein quantitation by considering the unique and shared peptides of both species.The results showed that SPA could provide more convenient and accurate protein quantitation in human-mouse mixed samples.Further validation on a pair of gastric PDX samples (one bearing FGFR2 amplification while the other one not) showed that our new method not only significantly improved the overall protein identification,but also detected the differential phosphorylation of FGFR2 and its downstream mediators(such as RAS and ERK)exclusively.The tool pdxSPA is freely available at https://github.com/Li-Lab-Proteomics/pdxSPA.
KEYWORDS Patient-derived xenograft model;Label-free;Shared peptide;FGFR2 amplification;Biomarker
Introduction
Patient-derived xenograft (PDX) is an animal model popularly used in cancer research,in which a patient’s primary tumor tissue is engrafted directly into an immunodeficient mouse.Compared with cell line-based xenografts,PDX could better maintain the fidelity of original tumors,including heterogeneity and tumor microenvironment [1-6].PDXs can resemble the drug-sensitivity patterns of the patients from which they derive,which may be used to predict clinical outcomes[5,7-10].
With the development of mass spectrometry (MS),cancer proteome analysis becomes popular in precision medicine,providing quantitative measurement of proteins and methodology to complement genomics [11-17].Meanwhile,PDX sample is gradually used for proteome analysis,which provides a new view for cancer research[16,18-23].In an integrated omic analysis of lung cancer,Li et al.[20]carried out genomics,transcriptomics,and proteomics analyses of 11 groups of non-small cell lung cancer(NSCLC)samples,and discovered that the patterns formed by metabolism proteins were highly recapitulated between primary tumors and PDXs by unsupervised clustering.In addition,Huang et al.[19]performed quantitative proteome and phosphoproteome profiling across 24 breast cancer PDX models,and identified multiple druggable protein events that were unique from the genome data,demonstrating the ability of MS-based PDX proteomics to identify therapeutic targets.
It should be emphasized that when the patient tumor tissue is engrafted to an immunodeficient mouse,the endothelial cells and fibroblasts from the host mouse take the place of human stromal components[24].Hence,the PDX sample is a mixture of human cancerous tissue and murine tissue.The percentage of human-originating cells varies among different PDX tissues,and it is not easy to be determined,especially for MS-based proteomics analysis.The bottom-up strategy is the most common way for proteome profiling by MS,which means that the tissue is digested into peptides prior to MS analysis [25].Due to the high homology of human and mouse proteins,a relatively high proportion of peptide sequences are shared by human and mouse.Therefore,the species of these peptides cannot be unequivocally determined,which may even lead to the misinterpretation of some mouse-unique peptides as human peptides.Some studies using PDX model perform database searching against human proteome sequences alone,such as an integrated omics analysis in lung cancer[20].Although MS data have been searched against human-mouse combined (HM) database in most current PDX proteome studies,only the human-specific peptides are considered for further analysis and quantification [18,19].However,omitting the peptides shared by two species could lead to great information loss,and thus the sensitivity of protein identification decreases dramatically.How to assign the shared peptides to taxa and how to use these data in quantitation are still challenging in PDX proteome data translation.Recently,Saltzman et al.[26]reported an algorithm gpGrouper for peptide grouping and protein quantitation at gene product level,in which the shared peptides between two species are distributed based on unique peptide peak ratios.However,the quantitation of those proteins,in which all the peptides are shared by human and mouse,has not been addressed yet.Due to the high homology between human and mouse,these kinds of proteins comprise not a small proportion in the identified proteins by MS/MS.
In this study,using the lab-assembled mixtures of human and mouse cell lysates with different proportions as PDX sample mocks,we proposed and assessed a new peptide quantitation method named SPA (shared peptide allocation).The new method makes reasonable allocations of the human-mouse shared peptides to improve the MS data interpretation of PDX samples.We proved that both false positive and false negative peptide identification existed when we used the human protein database alone or used the HM database with human-unique(HU)peptides picked up.We compared different strategies assigning peptides to human and mouse through searching against the HM database.The comparison results suggested that SPA is a good choice to balance the sensitivity of protein identification and the precision of protein quantitation.Moreover,SPA can provide an accurate estimation of the overall mixing ratio of tumor cells in PDX samples.We did further test on a pair of gastric PDX samples,one bearingFGFR2gene amplification while the other one not.Via our new strategy,we identified 23% more proteins than those identified in the usual way.Especially,tens of proteins involved in vital signaling pathways were uniquely identified in our new strategy.The analysis results suggested that our new strategy is feasible and could help to improve the reliability and analysis depth of PDX proteome data.SPA is implemented by Python and is freely available at https://github.com/Li-Lab-Proteomics/pdxSPA.
Results
Construction of a proteome benchmark dataset
To simulate the protein lysate of PDX tissues,we artificially generated four human-mouse protein mixtures with different ratios as a benchmark dataset.We used AGS cells(a type of human gastric adenocarcinoma cell line) to represent human cancerous tissue in a PDX sample and used mouse fibroblast (MEF) cells to resemble the murine elements.The two lysates were mixed at different ratios(Figure 1).The percentages of human proteins in these four mixtures were 60%(SET60),70%(SET70),80%(SET80),and 90% (SET90).Equal amounts of mixtures were digested by trypsin and then fractionated via house-packed C18tip into six fractions.Four samples were manipulated in parallel to reduce random effects.Technical replicates of MS analysis were performed.
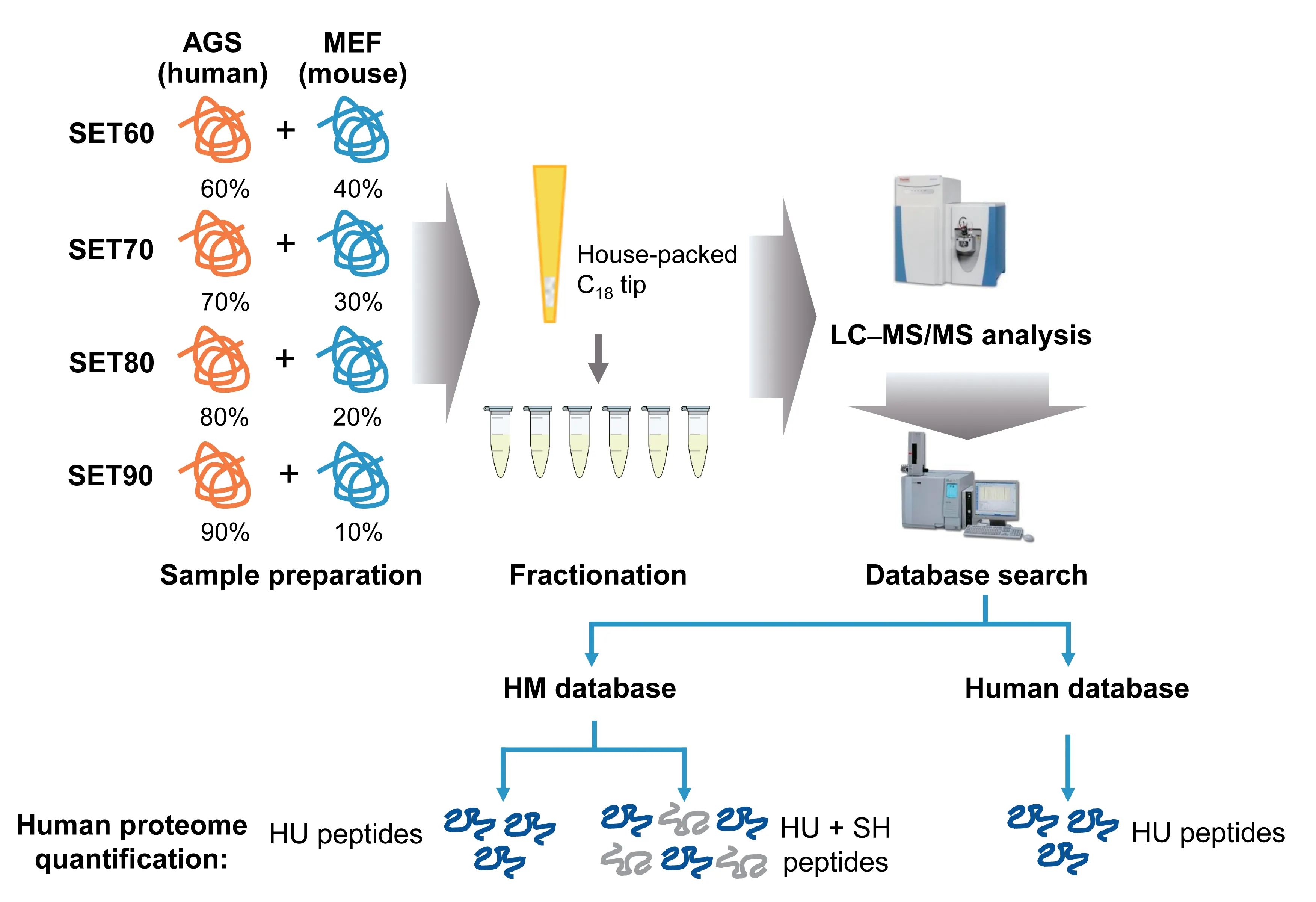
Figure 1 The construction of standard testing sets and the strategy of human proteome quantitation for PDX model
Peptide identification with different searching databases
To evaluate database selection in proteomics analysis in PDX samples,we compared the peptide identification results using HM database with the results using human database alone.All acquired MS/MS data files (.raw)of four standard testing sets were processed with MaxQuant based on Andromeda search engine.Homo sapiensandMus musculusprotein databases from UniProt were combined to form a new cross-species database(HM database).Then we performed peptide identification of the four mixture samples by searching against human database alone or against HM database.All parameters were the same except for the database selection.The false discovery rate (FDR) cutoffs of peptide and protein were set as 0.01.
We classified the identified peptides searched against HM database into three categories:1)human-unique(HU):amino acid sequences existing in human proteins uniquely;2)human-mouse shared(SH):amino acid sequences shared by human and mouse proteins;3) mouse-unique (MU):amino acid sequences uniquely mapped into mouse proteins but not present in humans.For a given MS/MS spectrum identified as a human peptide sequence against human database,it is possible to be explained as a SH or MU peptide with higher confidence when searching against HM database.
Based on the number of the peptide spectrum matches(PSMs)listed inTable 1,we found that approximately 60%PSMs,which had been assigned to human using human database alone,were SH ones with higher confidence.More importantly,there were hundreds of spectra of MU peptides“mismatched”to human peptide sequences in each dataset.Taking the results of SET60 as an example,we found that 992 spectra from the results against human database were re-assigned as MU peptides more confidently(Table S1).Amismatched example was shown inFigure 2.The spectrum(scan No.3029) in SET60 was identified as GAQEVGER when we used human database alone(Figure 2A).However,when we used HM database,this spectrum was identified as QGAEIDGR with higher match score,and was MU(Figure 2B).
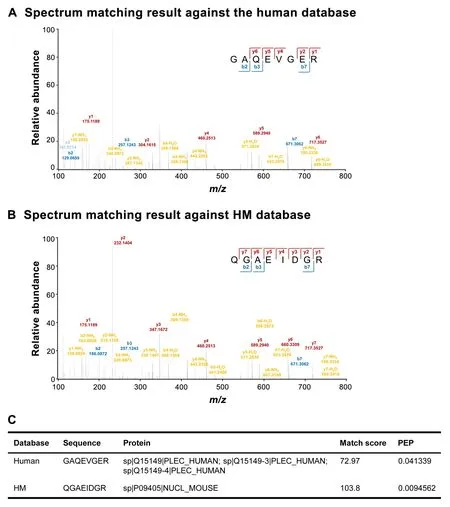
Figure 2 A spectrum example in human database and HM database
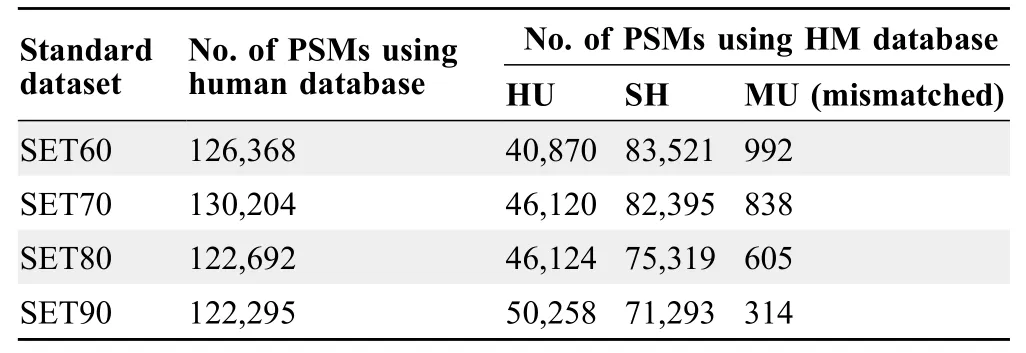
Table 1 No.of PSMs identified using different protein databases
Obviously,for protein quantitation of PDX samples,the indistinguishable and mismatched spectra will lead to biases and errors for quantitation if human protein database rather than HM database was used.Thus,we performed a further peptide identification against HM database.On average,56,395 peptides were identified in each sample.The proportions of HU,MU,and SH peptides in four testing sets are shown inFigure 3.With the increase of the percentage of human proteins in each sample,the number of identified HU peptides increased also,while the number of MU peptides decreased.When looking at the SH peptides,we found that more than half of the identified peptides were shared by human and mouse.Severe information loss could not be avoided if we only use HU peptides and discard SH peptides directly in protein quantitation.However,how to use these SH peptides remains a problem.
New algorithm for accurate protein quantitation of PDX models
A major challenge of protein quantitation in PDX models is to assign the shared peptides between human and mouse appropriately.Here we describe a new method named SPA for quantifying the MS data from the PDX models,especially focusing on the quantitation of the SH peptides.
We made descriptive statistics of the standard testing sets and then found that the ratio of the sum of HU peptide intensity (IHU) to the sum of MU peptide intensity (IMU)could best mimic the prior mixing ratio in each of standard testing sets.We assumed that the SH peptides could be allocated based on this ratio.Therefore our strategy is as follows:1)sort the peptides into three categories:HU,MU,and SH;2)calculate the individual ratioriof HU intensity to MU intensity for each protein,as well as the overall ratiorof the sum of HU intensity(IHU)to the sum of MU intensity(IMU)in the sample,representing the overall ratio of human proteins to mouse proteins in mixture.The individual and overall human-to-mouse ratios have the following form:ri=IHU/IMUandr=Sum(IHU)/ Sum(IMU);3) allocate the intensity of SH peptides according to the human-to-mouse intensity ratio.Finally,for a given human protein,its intensity equals HU peptide intensity (IHU) plus the human part from the SH peptides.The overview of the strategy is shown inFigure 4A.
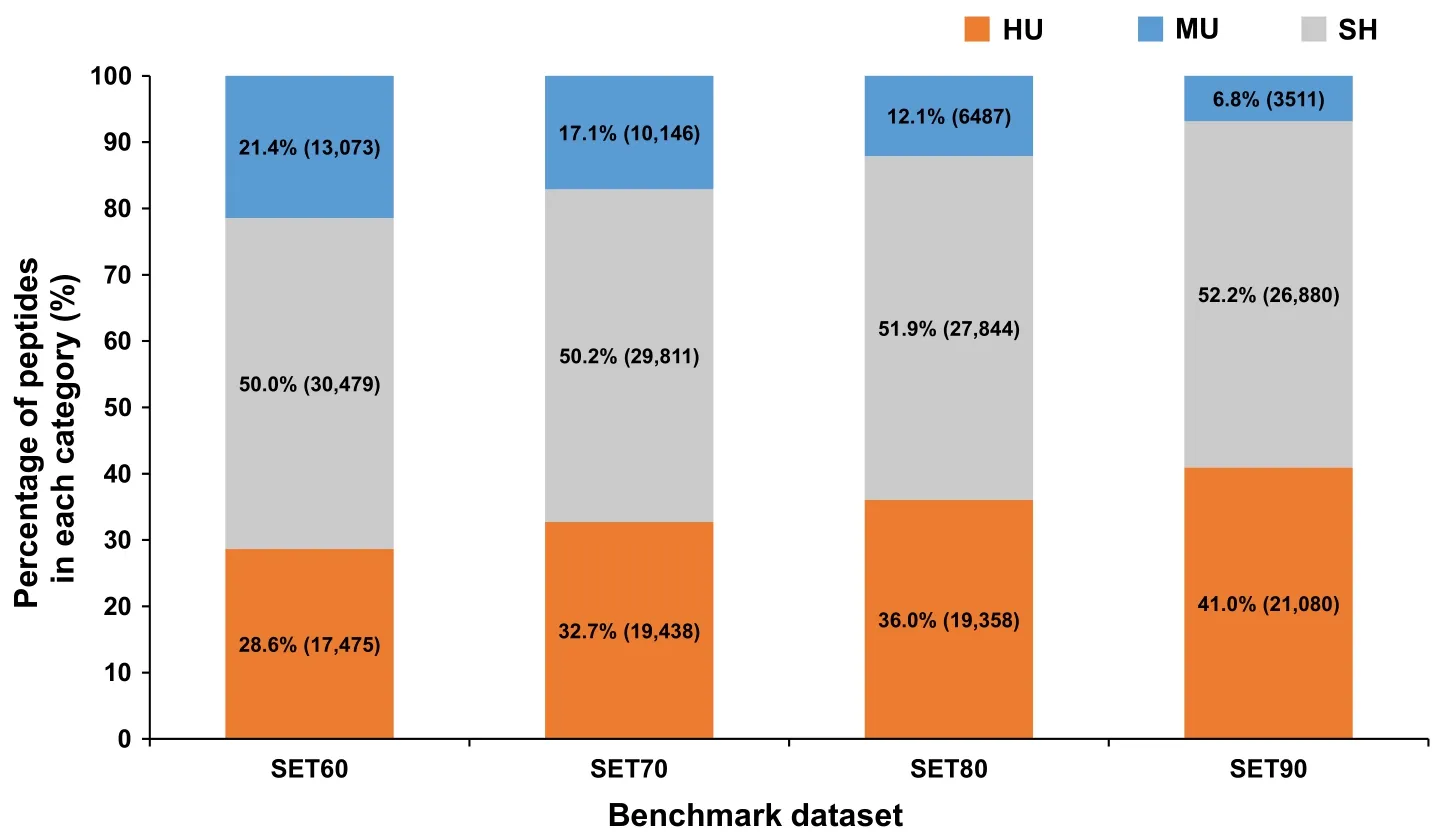
Figure 3 The proportions of HU,MU,and SH peptides in total peptide identification against HM database
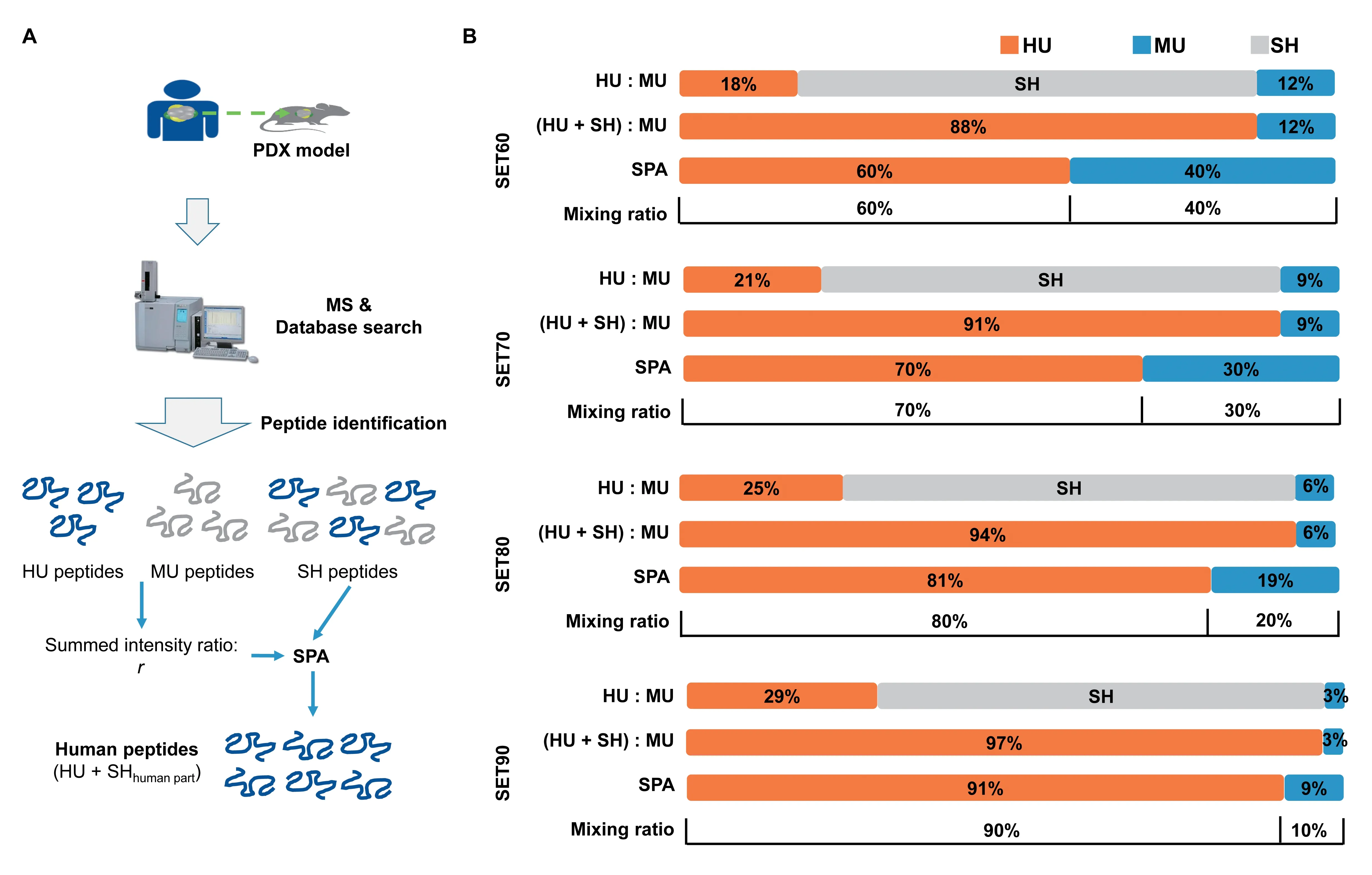
Figure 4 Strategy for PDX MS data quantitation
Through our new strategy,the overall human-to-mouse ratios in four testing sets were 60%:40%,70%:30%,81%:19%,and 91%:9%.Compared with our set ratios,the deviations were all within 1.5% (Figure 4B).It was clear that the ratio of the sum of HU intensity to the sum of MU intensity fitted the theoretical mixing ratio well.
Next,we assessed SPA about the recall in protein identification and the precision in protein quantitation.We selected all the gene-unique peptides,which matched to a single gene.Then we quantified proteins by their respective gene symbols with the sum of the intensity of gene-unique peptides,in which about 58% peptides are SH peptides.Presently,the most common strategy for PDX proteome data analysis is assembling proteins using HU peptides.Our new strategy is to assemble proteins using not only HU peptides but also SH peptides.In four testing sets(SET60,SET70,SET80,and SET90),3713,4008,4039,and 4270 proteins were separately identified via HU strategy,while we got 5508,5605,5530,and 5568 proteins using SPA.This result suggested that our new strategy SPA can reduce information loss and increase the sensitivity of protein identification significantly.
We evaluated the quantitation performance of SPA by comparing the expression of proteins identified in SET60 and SET90 datasets.After total intensity normalization,the theoretical ratio of a protein’s intensities in SET90vs.SET60 should be 1.5(Log21.5 ≈0.58).We calculated and plotted the ratios of protein intensity in two datasets(Figure 5A-E).Here we tested three strategies to assign the SH peptides.The first one(labeled as X-SPA)was to assign the SH peptides based on the individual human-to-mouse ratiori,which can vary significantly among different proteins.Only for proteins having both HU and MU peptides,all the SH peptides belonging to the certain protein were allocated at this individual ratio.This strategy is similar to gpGrouper[26].The second one(labeled as S-SPA)was to assign the SH peptides using X-SPA if both HU and MU peptides were identified;otherwise,assign the SH peptides based on the overall human-to-mouse ratior.The third one(labeled as SPA) is a simplified way,by which the SH peptides were assigned based on the overall ratiorin the quantitation of all proteins.Moreover,we performed comparisons with the other two existing methods for assigning the SH peptides.One was using HU peptides only in protein quantitation(labeled as HU);the other one was to assign all SH peptides to human(labeled as HU+SH).As shown in Figure 5A-E,we found that SPA and S-SPA showed better performance than the others as the median ratios of these two methods are quite close to the theoretical value 0.58.The standard variation(SD)of ratio estimation in SPA was the smallest,indicating that SPA is more stable for protein quantitation.Using SPA and S-SPA,more SH peptides were used for protein assembly,and therefore the number of quantifiable proteins increased in comparison with HU and X-SPA strategies(Figure 5F).
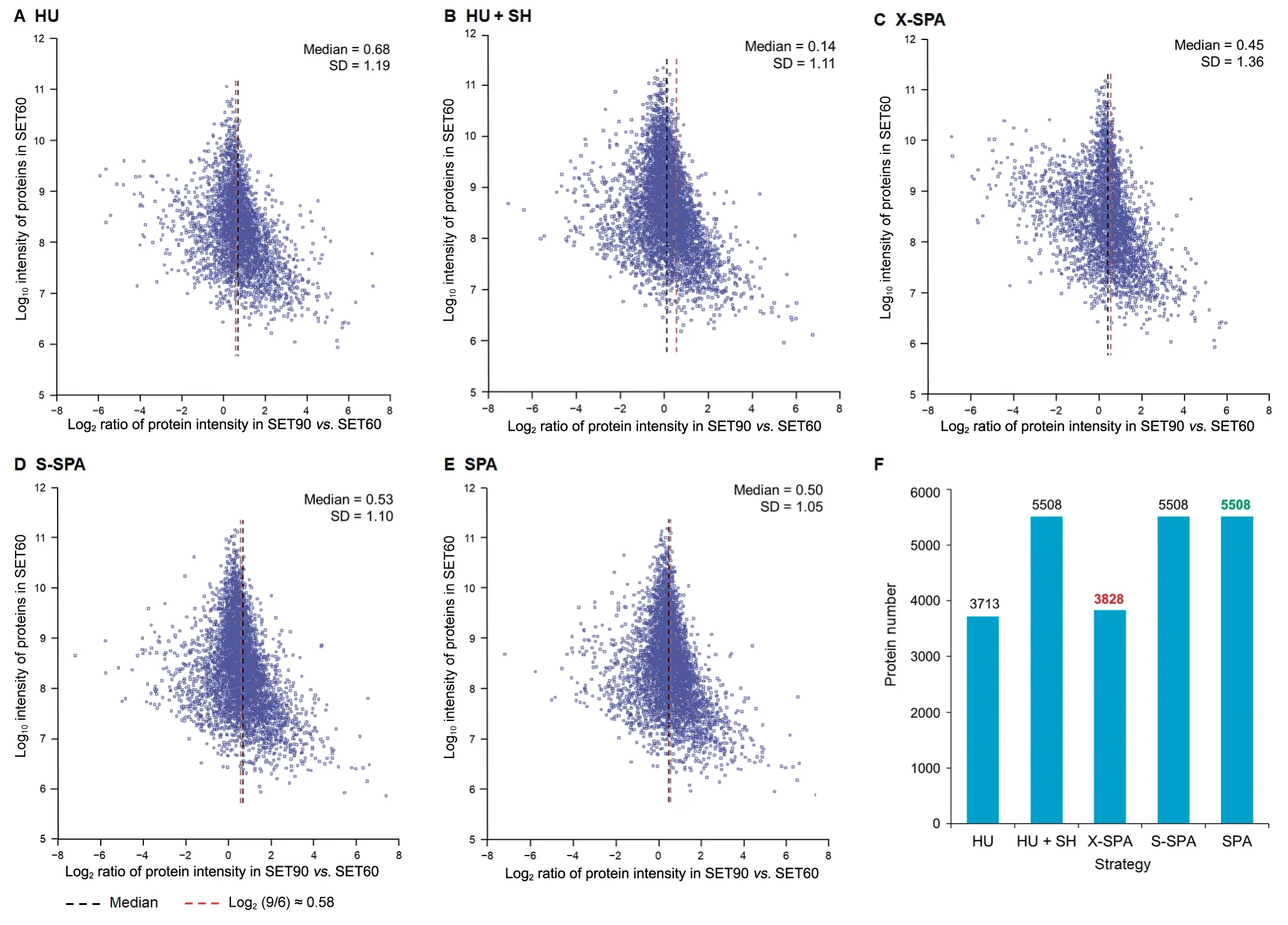
Figure 5 Comparison and evaluation of five quantitation strategies
In conclusion,among these five strategies shown in Figure 5,both SPA and S-SPA are acceptable in the accuracy and stability of ratio estimation.Therefore,SPA would be a better choice.
Validation on real PDX samples
In addition to the benchmark dataset,we further validated our new strategy for peptide and protein quantitation in real complex PDX data.FGFR2amplification in gastric cancers shows an association with poor prognosis,and it leads to the expression change of protein FGFR2 and the activation of its downstream mediators[27-31].Here,we analyzed a pair of gastric PDX samples derived from two patients,one bearingFGFR2amplification while the other withoutFGFR2amplification (control).
Label-free quantitation was used when we searched MS raw data against HM database.In total,89,936 peptides were identified.Again,we sorted these peptides into three categories:HU peptides (n=37,967),MU peptides (n=12,725),and SH peptides (n=39,244).The ratios of Sum(IHU) and Sum(IMU) in case and control samples were 1.86 and 1.78,respectively,which means that the purities of these two PDX tumor samples were just around 65%.To assemble the proteins,we selected the gene-unique peptides for protein quantitation.Finally,we got 7336 proteins using SPA,while we got 5940 proteins using HU peptides only in protein quantitation (the HU strategy mentioned above)(Figure 6A,left panel).Our new strategy increased the number of total identified proteins by 23.5%.We used the lab-available antibodies to test the expression levels of three proteins in two PDX tissue lysates,including PTEN,KRAS,and MEK1(also known as MAP2K1).These proteins were identified by our new strategy SPA,but could not be detected by the HU strategy (a traditional method).The following-up Western blot result showed that these proteins were indeed expressed in these PDX samples (Figure S1).In addition,these proteins had higher expression levels inFGFR2amplification sample than in the control sample,which was consistent with our qualification result via SPA(Figure S1;Table S2).With concern about the bias to lower intensity in peptide identification that possibly led by our strategy,we plotted the fold changes(FGFR2amplificationvs.control) and the average intensities of all identified proteins using the HU and SPA strategies.We found that the distribution patterns were quite similar (Figure S2).
Next,we analyzed the differentially expressed proteins in theFGFR2amplification sample compared to the control sample,and set 3-fold change as a relatively strict standard.According to the Venn diagram,we found that most differentially expressed proteins (>3-fold change or <0.33-fold change) could be identified via both strategies (Figure 6A,left panel).Of importance,among 1396 ‘additionally obtained’ proteins by SPA,267 proteins were up-regulated in theFGFR2amplification sample while 238 proteins were down-regulated (Figure 6A,left panel).Further enrichment analysis showed that up-regulated proteins were involved in several key biological processes including signal transduction and extracellular matrix organization (Figure 6A,right panel).Proteins such as MEK1,SOS1,and TGFBR2,were among this list.The enrichment analysis also suggested that down-regulated proteins participated in transcription regulation and glutamate secretion(Figure 6A,right panel).The gene set enrichment analysis also showed the strength of our new strategy,in which the important signaling pathways were detected with much higher sensitivity(Figure S3).
The phosphorylation level would be changed due toFGFR2amplification.Based on the characteristic of our PDX samples,we additionally analyzed the global phosphoproteome data and obtained 916 phosphorylated proteins uniquely identified via our new strategy.We took protein FGFR2 and its downstream mediators as an example (Figure 6B).The phosphorylation of several proteins such as GRB2 and ERK1/2 were exclusively identified and quantified by our new strategy.Of note,we found that protein ERK1/2 was activated inFGFR2amplification sample,which was consistent with the reported characteristic ofFRFG2-amplified gastric cancer[32].This result further demonstrate the power of our new strategy to rescue more peptide information and provide more accurate protein quantitation in PDX model.
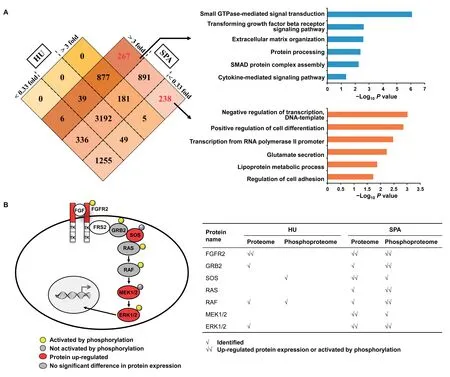
Figure 6 Protein identification and quantitation of two gastric PDX samples via two strategies
Discussion
In this study,we presented a new strategy for peptide/protein quantitation of PDX proteome data in order to address the existing problems and improve the precision of data analysis.We started with a distinctive experiment design that we artificially created a series of human and mouse protein mixtures with known percentages of human proteins.After comparing and evaluating several strategies,we established and recommended our new quantitation strategy SPA.Our results showed that no obvious quantitation bias was observed in SPA.Also this strategy helped to increase quantifiable proteins by near 35%.The calculated human protein ratios by SPA matched the prior ratios well,suggesting the reliability of our strategy.Additionally,compared with the previous way by which only HU peptides were retained,our method rescued some proteins including the SH peptides with relative high quantitation accuracy,and reduced information loss.Our results strongly suggested that the peptides shared by human and mouse should not be omitted.
In the evaluation of SPA’s performance in protein quantitation,we made a comparison of five potential methods.For X-SPA,it is the most theoretically accurate method,as the SH peptides are allocated at the exact ratio of each protein.However,from our scatter plot,we found that the ratio was underestimated and the variation was relatively large.As the human-to-mouse ratio of each protein should be calculated,missing or inaccurate intensity measurement of low abundant or other certain peptides may impact this ratio’s calculation.
SPA is a reliable and feasible method for data processing.Also,with the usage of most SH peptides,it could increase the number of the identified and quantifiable proteins,and also improve the precision in protein quantitation.Since a human-to-mouse ratio was used in SPA as a global scalar for the allocation of SH peptides,some proteins,which share all of the identified sequences between human and mouse but are only expressed in mouse tissues in fact,will unavoidablybe considered as “human proteins/peptides”.Using HU peptides alone for protein assembly can avoid this problem.However,such method(i.e.,the HU strategy in our study)would lead to drastic loss of those truly human expressed proteins,which share the same sequences to their corresponding mouse proteins.Moreover,as shown in our data,these proteins,whose identified peptide sequences are shared by human and mouse,consist of a significant 30%of all the proteins identified(Figure 5F);simply ignoring them would lead to a huge information loss and inefficient usage of the proteomics data.Based on the nature of proteome,we believe that these fake human proteins consist of a very small proportion.The impact of these proteins would be even more negligible in a typical PDX model study as people usually only consider the differentially expressed proteins between the PDX samples.
At last,we applied the new strategy to a gastric tumor PDX sample withFGFR2amplification and a paired negative control respectively.The improvement in protein identification and quantitation demonstrated the power of the new method very well,which keeps the consistence with the prior knowledge aboutFGFR2amplification.However,more biological replicates should be added if we want to justify the biological changes and explore the underneath molecular mechanism of a specific PDX model.
In summary,the analyses in both the benchmark dataset and the real PDX data suggest that our strategy has several advantages for PDX proteomics analysis.First,SPA is helpful to correct the impact of purity difference in different samples,but also to rescue more proteins in identification.Second,compared with the traditional strategy using HU peptides only and other peptide-allocation methods,our approach shows higher accuracy in protein quantitation,and is more user-friendly in the calculation.From a systematic view,our method shows advancement in protein identification and quantitation,which would further help efficient PDX data mining.
Materials and methods
Sample preparation and protein extraction
Mixture of human and mouse proteins
AGS cells (a type of human gastric adenocarcinoma cell line) and MEF cells from American Type Culture Collection (Rockville,MD) were separately cultured in DMEM medium(Catalog No.10569-010,ThermoFisher Scientific,Waltham,MA)supplemented with 10%fetal bovine serum(Catalog No.04-121-1A,Biological Industries,HaZafon,Israel).When cell confluence reached about 90%,cells were digested by trypsin and collected.After pre-cold PBS wash,cells were suspended in lysis buffer (8 M Urea in 100 mM NH4HCO3,protease inhibitor cocktail,pH 8.0)and stayed on ice for 30 min.Sonication was used to disrupt DNA aggregation.Both lysates were centrifuged at 21,130gat 4°C for almost 10 min.Then the supernatants of AGS and MEF cells were transferred to the clean centrifuge tubes,respectively,and protein concentrations were determined by bicinchoninic acid (BCA) assay (Catalog No.P0010,Beyotime Biotechnology,Shanghai,China).
For a series of human-mouse protein mixtures,we firstly diluted both of the AGS and MEF lysates to the concentration of 5 mg/ml.For SET60,24 μl AGS lysate and 16 μl MEF lysate were mixed.Similarly,SET70 consists of 28 μl AGS lysate and 12 μl MEF lysate;SET80 consists of 32 μl AGS lysate and 8 μl MEF lysate;SET90 consists of 36 μl AGS lysate and 4 μl MEF lysate.
PDX samples
PDX animal samples were generated from primary gastric cancer tissues.The samples were provided and validated by Crown Bioscience (Beijing,China) in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health.Two cryopulverized PDX tissues (one bearingFGFR2amplification and the other one not) were used for further proteome analysis.These two PDX samples were washed with pre-cold PBS twice and then mechanically sectioned into tiny fragments before lysis.Tissues were suspended in lysis buffer and stayed on ice for about 30 min.Then the lysates were sonicated for 5 min(3-s sonication and 5-s interval as a cycle)for complete lysis.The supernatants were transferred to new Eppendorf tubes after centrifugation at 21,130gat 4°C for almost 10 min,and protein concentrations were determined by BCA assay.The paired PDX samples were treated in parallel to eliminate manipulation error.
In-solution trypsin digestion
Cell lysates were incubated with 5 mM dithiothreitol(DTT)at 56°C for 30 min.After reduction,the lysates were alkylated by 15 mM iodoacetamide (IAA) at room temperature in the darkness for 30 min,and quenched with 30 mM cysteine for another 30 min.The protein solutions were diluted in 2 M Urea with 100 mM NH4HCO3(pH 8.0)and then digested with Sequencing Grade Modified Trypsin(Catalog No.V5111,Promega,Madison,WI)at 1:50(w/w)trypsin-to-protein ratio at 37°C for 16 h.To complete the digestion cycle,trypsin was then added at 1:100 (w/w)trypsin-to-protein ratio at 37°C for another 4 h.Peptide desalting was conducted via SepPak C18cartridges(Catalog No.186006325,Waters,Milford,MA) and the eluted solution was vacuum dried before fractionation.
Peptide fractionation
The human-mouse protein mixtures were fractionated using house-packed C18columns.C18beads were resuspended in acetonitrile (ACN) and transferred onto top of previously equilibrated C18StageTips,and then washed by 150 μl buffer A [98% H2O,2% ACN,pH 10 (adjusted by NH4OH)].Tryptic peptides were dissolved in 80 μl buffer A and loaded onto the C18beads twice.The flow through was retained and marked as E1.The combined peptides were sequentially eluted with 4%,8%,12%,16%,20%,24%,28%,32%,40%,60%,and 80% buffer B [98% ACN,2%H2O,pH 10 (adjusted by NH4OH)],and marked as E2 to E12,respectively.The eluted peptides were combined to form six fractions.
For PDX samples,the desalted peptides were fractionated by Agilent 1100 HPLC system with XBridge C18column (4.6 mm × 100 mm,130 Å pore size,3.5 μm particle size;Catalog No.186003033,Waters).Peptides were separated with a gradient of 2%-100%buffer B(98%ACN,2% H2O,pH 10.0) at a rate of 0.6 ml/min in 60 min.The peptides were combined into ten fractions.Each fraction was vacuum dried in SpeedVac (ThermoFisher Scientific)for further MS analysis.
LC-MS/MS data acquisition
The fractions of four human-mouse protein mixtures were analyzed on Q Exactive (ThermoFisher Scientific).Labmade reversed-phase C18pre-column (4 cm length,75 μm ID,5 μm particle size) and C18analytical column (16 cm length,75 μm ID,3 μm particle size) were used for online desalting and peptide separation.For each fraction,peptides were dissolved in Mobile phase A[H2O with 2%ACN and 0.1%fatty acid(FA)]and then loaded onto the pre-column.Peptides were separated with Mobile phase B (ACN with 2%H2O and 0.1% FA) under a linear gradient from 5% to 35%for 30 min and from 35%to 80%for 10 min at a flow rate of 300 nl/min.The eluted peptides were ionized via nano-electrospray ionization(NSI)source and introduced to Q Exactive.The resolution of intact peptides withm/z350-1600 for Orbitrap was set as 70,000 atm/z200.The 16 most intense ions,whose intensity was higher than 2×104,were further selected and fragmentized by higher-energy collision dissociation (HCD) with normalized collision energy of 28%.The resolution of ion fragments was set as 17,500 atm/z200.The dynamic exclusion duration was 60 s.The isolation window was set asm/z2 and automatic gain control (AGC) was on.Technical replicates were performed.
The fractions of gastric PDX samples were analyzed on Fusion (ThermoFisher Scientific).Full MS spectra (fromm/z350 tom/z1300) were acquired with the resolution 120,000 atm/z200 in profile mode.For full scan,AGC targets were set as 5.0×105within the maximum injection time of 50 ms and for MS/MS scan,AGC targets were set as 7.0×103within the maximum injection time of 35 ms.Data dependent mode was set as top speed and the most intense precursor ions were fragmented by HCD with normalized collision energy of 32%.MS/MS spectra were detected by ion trap and ion trap scan rate was set as rapid.Dynamic exclusion duration was set to 60 s.The fractions of these two samples were subjected to MS sequentially.Two technical replicates for each sample were conducted.
Data processing
Database searching
All the acquired MS/MS raw data were processed with MaxQuant (version 1.5.3.8) based on Andromeda search engine[33].
Peptides were identified against the HM database which contained the sequences of UniProt human (proteome ID:UP000005640,last modified in Oct,2015) and UniProt mouse (proteome ID:UP000000589,last modified in Oct,2015),and enabled contaminants and reversed sequences.For comparison,some data were additionally searched against UniProt human database.The search results were filtered to a 1% FDR at the peptide and protein levels.Trypsin/P was specified as the digestion enzyme,and maximum missing cleavage was set as 2.Precursor error tolerance was set as±10 ppm with fragment ion±0.02 Da for Q-Exactive and fragment ion ± 0.5 Da for Fusion.Modification parameters were as follows:carbamidomethyl(C)as the fixed modification,oxidation(M)and acetylation(Protein N-terminus) as variable modifications.
For each standard testing set,as two technical replicates were conducted under the same MS condition,the raw data were labeled as experiment “R1” and “R2”,respectively.Match between runs was ticked and set with a minimum window of 0.7 min.
For PDX samples,label-free quantitation mode was chosen with default parameters and “match between runs”was set with a minimum window of 0.7 min.
The files “peptide” and “evidence” generated from software MaxQuant were used for subsequent analysis.Data were processed with R language.For “peptide” file,the peptides labeled as contaminant or reverse were removed before other procedures.Peptide intensity was used for peptide and protein quantitation.
Comparison of peptides identified in human database and HM database
We used the spectra ID and score in “evidence” file produced by MaxQuant for subsequent processing.We regarded a spectrum as a MU peptide in HM database,only if its search score is higher than the corresponding score in human protein database.
Protein assembling and quantitation
Gene-unique peptides which were matched to only one gene were selected for protein assembly.The intensities of all respective peptides were summed up as protein intensity.As technical replicates were performed,we took the average to represent the protein expression value.The protein expression values were normalized by the summed intensity of each sample that reflects the total abundance(Table S2).
PDX sample analysis
Column “intensity” of each peptide in “peptide.txt” was used for peptide quantitation and protein assembling.Total intensity normalization was performed to adjust the difference of total peptide amounts in two samples (the same strategy used in standard testing set normalization).As for the phosphorylation data,the intensity of phosphopeptides was normalized to the abundance of the parent protein,which reflects the changes in relative phosphorylation of the protein without confusion by changes in expression of the protein itself [13].Missing values were filled with minimum number 100,000.Proteins were assembled via the HU strategy or SPA.The fold change was calculated for each protein via the average intensity in theFGFR2amplification sample dividing by that in the control sample.Then we chose proteins with more than 3-fold change or less than 0.33-fold change for subsequent enrichment analysis.Enrichment analysis was performed using DAVID 6.8 tools with the totalH.sapiensgenome as the background [34,35].
Code availability
The tool pdxSPA is freely available at https://github.com/Li-Lab-Proteomics/pdxSPA.
Data availability
All MS raw data have been deposited to the PRIDE [36]partner repository(ProteomeXchange:PXD008611)which are publicly accessible at http://proteomecentral.proteomexchange.org/cgi/GetDataset.
CRediT author statement
Xi Cheng:Methodology,Software,Validation,Data curation,Writing -original draft,Writing -review &editing.Lili Qian:Validation,Investigation,Writing -original draft,Writing-review&editing,Visualization.Bo Wang:Methodology,Formal analysis,Validation,Data curation,Writing -original draft,Visualization.Minjia Tan:Conceptualization,Investigation,Resources,Supervision,Writing-original draft,Writing-review&editing,Project administration,Funding acquisition.Jing Li:Conceptualization,Methodology,Resources,Supervision,Writing-original draft,Writing-review&editing,Project administration,Funding acquisition.All authors have read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This study was supported by the Special Project on Precision Medicine under the National Key R&D Program of China (Grant No.2017YFC09066600),the National Natural Science Foundation of China (Grant Nos.31871329,31670066,and 31271416),the National Science &Technology Major Project“Key New Drug Creation and Manufacturing Program”,China (Grant No.2018ZX09711002-007),and the Natural Science Foundation of Shanghai,China (Grant No.17ZR1413900).We thank the High Performance Computing Center(HPCC)at Shanghai Jiao Tong University for the computation.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2019.11.016.
ORCID
0000-0002-8120-9409 (Xi Cheng)0000-0002-5882-1878 (Lili Qian)0000-0003-0975-9120 (Bo Wang)0000-0002-6784-9653 (Minjia Tan)0000-0003-4602-3227 (Jing Li)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- RePhine:An Integrative Method for Identification of Drug Response-related Transcriptional Regulators
- NOGEA:A Network-oriented Gene Entropy Approach for Dissecting Disease Comorbidity and Drug Repositioning
- DeepCAPE:A Deep Convolutional Neural Network for the Accurate Prediction of Enhancers
- The Genome Sequence Archive Family:Toward Explosive Data Growth and Diverse Data Types
- Genome Warehouse:A Public Repository Housing Genomescale Data
- REVA as A Well-curated Database for Human Expressionmodulating Variants