mtDNA C11777A突变所致Leigh综合征2例报道
2021-03-24刘晓鸣
桑 艳,刘晓鸣,岳 璇
Leigh综合征又称为亚急性坏死性脑脊髓病,是由遗传缺陷引起线粒体代谢酶异常,ATP合成障碍、能量来源不足,导致严重神经肌肉损伤,以脑基底节、脑干、脊髓、小脑等部位对称性坏死病变为病理表现。由于病人症状多样,发病率低,临床对本病认识不足,常存在误诊漏诊。现分析2例同胞兄弟Leigh综合征病例如下。
1 资 料
病例[1] 患儿,男,7岁7个月,主因“吐词不清3个月,吞咽困难半个月”于2016年11月21日入院。3个月前家长发现患儿说话含糊不清,声音小,未予重视,半个月前出现进食固体食物困难,逐渐发展成流质饮食困难,伴有呛咳、流涎。患儿既往发育稍落后于同龄儿童,1岁10个月行走,2岁叫“爸爸、妈妈”,3岁会说简单句子,目前上小学2年级,成绩中等。婴儿时发现有左眼斜视。入院查体:意识清楚,呼吸浅快,偶有叹气样呼吸,步态正常,不能快跑,双足跳不稳,计算能力减退,说话声音小,后背部毛发多,流涎明显,面部表情僵硬,转头及耸肩无力,左眼外斜视,双眼眼球震颤(+),咽反射消失,心肺查体无特殊,四肢肌张力稍高,肌容积正常,肌力Ⅳ级,膝反射活跃,巴氏征(-)。辅助检查:血乳酸5.7 mmol/L,血氨34.9 μmol/L,肝肾功能、血电解质、心肌酶均正常。血、尿遗传代谢筛查均为阴性,心脏彩超正常,脑电图正常,2016年11月21日血气分析:pH 7.394,氧分压(PO2)68.5 mmHg(1 mmHg=0.133 kPa),二氧化碳分压(PCO2)47.6 mmHg,HCO3-29.0 mmol/L,碱剩余3.7 mmol/L,2016年11月23日血气分析:pH 7.314,PO2122 mmHg,PCO267.2 mmHg,HCO3-34.1 mmol/L,碱剩余7.0 mmol/L,脑脊液常规+生化正常,脑脊液寡克隆带阴性。自身免疫性脑炎抗体均为阴性,血细胞沉降率、血狼疮全套均正常,肌电图:四肢近端肌肉运动单位时间缩短,四肢神经运动神经传导测定和感觉神经传导正常。头颅MRI:双侧尾状核体部、脑干可见多发片状稍长T1稍长T2稍高Flair信号,脑干异常信号呈轻度强化,脊髓MRI:未见异常,详见图1。
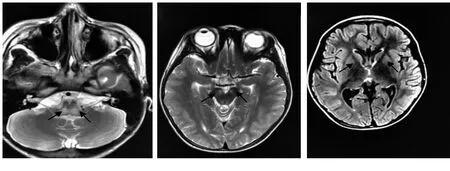
图1 病例[1]头颅MRI图像
病例[2] 病例[1]的弟弟,4岁,自幼发育落后,查体:意识清楚,呼吸平稳,体格匀称,步态正常,主动语言少,余查体未见异常。头颅MRI:小脑半球及脑干可见多发片状稍长T1稍长T2稍高Flair信号,详见图2。

图2 病例[2]头颅MRI图像
由于两兄弟均有相似的临床及影像学检查,诊断为Leigh综合征,给予补液,维生素等治疗,住院第3天病例[1]受凉后合并肺部感染,血气分析提示CO2潴留,转ICU治疗行辅助通气治疗,次日家长要求出院,出院次日患儿死亡。病例[2]患儿给予鸡尾酒疗法,门诊随访,目前出现时有叹气,不能吃固体食物。线粒体高敏感DNA测序报道2例患儿及患儿母亲均检测线粒体DNA变异(MT-ND4 m.11177C>G),符合Leigh综合征临床诊断。
2 讨 论
Leigh综合征是婴幼儿期常见的线粒体疾病之一,有研究证明,多种线粒体呼吸链复合酶、丙酮酸脱氢酶复合物及ATP合成酶缺陷均引起该病[1]。由Leigh于1951年首次报道[2],呈线粒体基因遗传、常染色体隐性遗传或连X连锁遗传。以脑基底节、脑干、脊髓、小脑等部位对称性坏死病变为特征。有研究资料显示,Leigh综合征中线粒体DNA(mitochondrial DNA,mtDNA)突变约占10%[3],属于母系遗传,核基因组DNA(nuclear DNA,nDNA)突变约占90%[4],遵循孟德尔遗传定律。张尧等[5]研究Leigh综合征,发现12例患儿存在mtDNA突变,最常见的是T10191C(3例),其次为A3243G(2例)、T8993G(2例),4例存在DNA突变,最常见的是SURF1(3例)。本研究中2例患儿突变基因为MT-ND4 m.11177C>G,该基因突变导致编码烟酰胺腺嘌呤二核苷酸(NADH)脱氢酶亚单位4(ND4)中第340位精氨酸变成丝氨酸,改变ND4空间构型,NADH脱氢酶活性降低,线粒体产能效率下降,ATP减少,长期能源不足导致神经元丢失和神经胶质增生,常累及基底节区和脑干。MT-ND4突变遵循母系遗传,具有阈值效应,病例[1]基因突变率为62.36%,母亲基因突变率为6.4%,因突变率低,无临床症状,病例[2]突变率为67.73%,发病年龄早于病例[1]。该突变位点少见,国外有5例报道[6-9],起病年龄7个月至67岁,方方等[10]基因确诊的35例Leigh患儿中1例是MT-ND4 m.11177C>A突变。
Leigh综合征临床表现多样,从无任何症状到严重神经系统异常均有可能,常见的临床表现包括眼球活动异常(78%)、呼吸障碍(69%)、肌张力低下(69%)、锥体束损害(61%)、运动障碍(58%)等[11]。Leigh综合征根据发病年龄分为新生儿型、经典婴儿型及少年型[12],成人型罕见。新生儿型起初多表现为吸吮、吞咽障碍及呼吸困难,之后逐渐出现异常眼运动、面肌无力等脑干症状,常早期死亡。经典婴儿型常于1岁以内起病,发病前精神运动发育多正常,发病后感染及高碳水化合物饮食可加重症状。少年型常在儿童期隐匿起病,逐渐出现共济失调、运动不耐受、眼震等临床症状,经过长时间静止期或缓慢进展后,10余岁时突然出现急性或亚急性恶化,迅速进展至昏迷及严重呼吸抑制,最终死亡。随着影像学技术发展,尤其是磁共振功能成像,从分子水平评估病变特征,双侧对称性基底节区和(或)脑干病变,称为Leigh典型改变,尤其是壳核受累普遍认为是Leigh综合征的特征,脑白质受累可见,表现为额叶、颞叶受累,也可为弥漫性脑室旁白质病变,少数病例甚至仅有白质受累或正常,而无基底节及脑干病变,因此,无典型基底节、脑干MRI表现,不能否定Leigh综合征诊断。方方等[10]研究35例基因确诊为Leigh综合征的患儿,MRI均累及脑干和(或)基底节区,其中27例(77%)累及脑干,24例(69%)累及基底节区,14例累及延髓,8例累及小脑。肖江喜等[13]研究32例Leigh综合征患儿MRI特点,其中8例常规MRI未见异常,63%累及深部核团,54%累及脑白质,磁共振波谱成像(magnetic resonance spectroscopy,MRS)对Leigh综合征诊断具有较高的临床价值。Leigh综合征患儿MRS常表现为Cho峰升高和NAA峰减低,在病变、脑脊液或看似正常的脑实质内可见乳酸双峰[13-16]。本研究中两例患儿突变基因相同,但磁共振累及部位不完全一致,病例[1]累及部位为双侧尾状核体部及脑干,病例[2]MRI主要累及小脑半球及脑干,并无病灶,两例患儿均未做MRS。由于Leigh综合征病发病率低,临床表现复杂及遗传异质性,诊断存在一定困难,既往Leigh综合征诊断多依靠死亡后病理分析或肌肉活检,随着医学影像学和分子遗传学发展,越来越多患儿可在生前诊断。1996年Rahman等[4]制定Leight综合征的诊断标准:进行性智力和(或)运动落后;有脑干或基底节损害症状;脑脊液或血乳酸增高;典型的影像或病理学表现,或同胞有典型病理表现,前3项加第4项中任意一项均可诊断为Leigh综合征。基因分析是Leigh综合征诊断与预防的重要手段,对疑似患儿应留取血样,以进行基因学诊断及分析。
Leigh综合征预后差,目前无有效的治疗措施,部分丙酮酸脱氢酶缺陷患儿,大剂量维生素B1及生酮饮食有一定效果。辅酶Q10、左旋肉碱、生物素、维生素B2、维生素B6、维生素C、维生素K等对电子传导障碍患儿有效[11,17-18],但剂量与疗效个体差异较大。患儿1岁内发病多在2岁左右死亡[19],晚发患儿(2岁以后)一般进展缓慢,可生存至10岁以上或成人[20]。
