红螯螯虾(Cherax quadricarinatus)卵黄蛋白原未知功能结构域(DUF1943)的原核表达及纯化鉴定
2021-03-22韦孜娜杨彦豪黄黎明韦秀颖卢天和黄光华杨慧赞
王 瑞,韦孜娜,吕 敏,杨彦豪,黄黎明,韦秀颖,卢天和,黄光华,杨慧赞*
红螯螯虾()卵黄蛋白原未知功能结构域(DUF1943)的原核表达及纯化鉴定
王 瑞1,韦孜娜1,吕 敏1,杨彦豪1,黄黎明1,韦秀颖2,卢天和1,黄光华1,杨慧赞1*
(1. 广西壮族自治区水产科学研究院,南宁 530021; 2. 广西大学动物科学技术学院,南宁 530005)
通过外源表达并纯化得到红螯螯虾()卵黄蛋白原(Vitellogenin,Vg)的DUF1943结构域片段,为开展其后续相关功能及机制研究奠定基础。根据GenBank数据库中公布的红螯螯虾卵基因序列(登录号AF306784.1),查找到DUF1943结构域的氨基酸序列(617—918aa),对其理化性质与基本结构等进行了理论评估,结合大肠杆菌密码子偏好对其进行了序列改造和密码子优化,采用全基因合成的方法得到了的基因片段,将其连接到Pet28a(+)内构建了Pet28a(+)-的融合表达载体,探索了该融合载体在大肠杆菌系统中的最佳表达参数,采用溶解法和上柱层析法对收获到的蛋白包涵体进行复性。全基因合成方法制备得到的目的基因为921 bp,成功构建了Pet28a(+)-的融合表达载体,诱导Pet28a(+)-表达的最佳条件为:当单克隆菌液OD600达到0.6 时,添加0.5 mmol·L-1的诱导剂IPTG,置于37 ℃条件下培养4 h。表达的DUF1943多肽以包涵体形式为主,在SDS-PAGE胶的35 kDa附近呈现特异性条带,收集包涵体进行破碎,经2 mol·L-1盐酸胍溶解和Ni-NTA层析后最终收获到的活性蛋白浓度为0.6 mg·mL-1。成功构建了Pet28a(+)-原核表达载体并优化了表达系统及蛋白复性方案,获得了DUF1943活性多肽,为进一步研究其生物功能及应用奠定了基础。
卵黄蛋白原;基因;原核表达;纯化;复性
卵黄发生是指各种卵黄物质的形成及其在卵母细胞中的积累,是卵母细胞发育成熟的必要前提,也是雌性甲壳动物生殖周期的决定性时期[1]。卵黄是由母体供应的,含有多种营养性物质成分,为虾类正在发育的早期胚胎和幼体提供所必需的能量物质。因此,卵黄的产生对于虾类的早期生长发育有着关键性的作用。Abdu等[2]分别提纯和鉴定了斑节对虾()、中国对虾()和罗氏沼虾()的卵黄蛋白(yolk protein),均为一种雌性的高密度脂蛋白(HDL)。Oberdörste证明了卵黄蛋白是胚胎发育的营养源[3]。
卵黄蛋白原(vitellogenin,Vg)是卵黄蛋白的前体,最初是Pan等[4]用于描述雌性昆虫血液中一种特殊淋巴蛋白,后来逐渐成为一些鱼类、禽类等性成熟的雌性脊椎动物的特异性血清蛋白的代名词[5-8]。卵生动物在雌激素诱导作用下,于卵巢以外的不同组织中(如海胆体腔细胞;鱼类、鸟类肝脏;甲壳动物滤泡细胞等)表达和合成卵黄蛋白原,经循环系统分泌运送到卵巢组织,在受体介导的内吞作用进入卵细胞,准确地表达了卵细胞的正常发育,为早期发育的胚胎组织提供了所需的脂肪、氨基酸、碳水化合物、磷和硫、维生素等功能性营养物质,促进卵细胞的生长分化[9-14]。对卵黄蛋白原的结构研究表明,卵黄原蛋白编码的3个结构域,它们分别是位于N端功能序列的结构域(vitellogenin-N)、位于C端的蛋白成分结构域(von Willebrand factor type D domain, vWD)以及富含磷酸化的结构域(domain of unknow function, DUF1943, DUF1944)[15-16]。这些结构域的分子结构在不同物种中,具有一定的保守性[17]。红螯螯虾()俗称澳洲淡水龙虾,是水产养殖业极具潜力的淡水经济虾类之一。卵黄发生过程中,消化酶和同工酶对卵黄的水解作用使其为红螯螯虾胚胎发育提供了重要的结构和营养性物质保障。近年来,对甲壳动物消化酶和同工酶的结构及功能研究多集中在幼体和成体的发育阶段[18-19]。而对于Vg的研究多集中在蛋白整体功能上,而具体针对其结构域的功能研究较少。结构决定功能,Vg蛋白结构域的差异正是其种间特异性的基础,因此对Vg蛋白的结构域进行独立的研究有利于更深入地认识和发现其功能和作用机制。
本研究根据红螯螯虾的Vg序列上的DUF1943结构域序列进行理论评估,结合大肠杆菌密码子的偏好性,对目的序列进行优化设计,采用全基因合成的方法克隆出了Vg的序列,并成功构建了Pet28a(+)-原核表达载体,诱导其在大肠杆菌感受态细胞BL21(DE3)中表达,进而纯化获得DUF1943蛋白,为进一步研究DUF1943的生物学功能奠定基础。
1 材料与方法
1.1 材料
大肠杆菌BL21(DE3)为本实验室保存。质粒Pet28a(+)、LB肉汤琼脂培养基、非预染蛋白Marker、预染蛋白Marker、非干扰型蛋白质浓度测定试剂盒、TMB显色试剂盒、IPTG均购自生工生物工程(上海)股份有限公司;DNA聚合酶、限制性内切酶、T4连接酶均购自Promega公司;其他试剂为进口或国产。
缓冲液A为pH 8.0的PBS;缓冲液B为8 mol·L-1Urea, 50 mmol·L-1Tris-HCl和300 mmol·L-1NaCl的混合液,pH 8.0;缓冲液C为50 mmol·L-1Tris,300 mmol·L-1NaCl,0.2 mmol·L-1PMSF和0.1% Triton X-100,pH 8.0;Binding buffer为50 mmol·L-1Tris,300 mmol·L-1NaCl,pH 8.0;Washing buffer为50 mmol·L-1Tris,300 mmol·L-1NaCl和20/50 mmol·L-1Imidazole,pH 8.0;Elution buffer为50 mmol·L-1Tris,300 mmol·L-1NaCl 和500 mmol·L-1Imidazole,pH 8.0。
JY88-II超声波细胞破碎仪(天津津立仪器设备科技发展有限公司);HZQ-F280全温度振荡培养箱(常州锐品精密仪器有限公司);GL-21M高速冷冻离心机(湖南湘仪离心机仪器有限公司)。
1.2 方法
1.2.1 DUF1943生物信息学分析 登录NCBI数据库(https://www.ncbi.nlm.nih.gov/ipg/?term=AF306784.1)查找到红螯螯虾(Cheraxquadricarinatus)基因的mRNA序列(登录号:AF306784.1),定位到其中DUF1943(617—918aa)结构域的基因序列,利用ORF finder(https://www.ncbi.nlm.nih.gov/orffinder)查找序列的开放阅读框并翻译其编码的氨基酸序列,将翻译的氨基酸序利用ExPAsy数据库(https://www.expasy.org/)的在线工具:ProtParam tool、ProtScale和InterProscan及TMHMM (http:// www.cbs.dtu.dk/services/TMHMM/)对DUF1943的亲水性、信号肽、跨膜域、基本结构等生物学信息进行分析和理论评估。利用在线平台Cell-PLoc-2 (http://www.csbio.sjtu.edu.cn/bioinf/Cell-PLoc-2/)选择真核生物系统(Euk-mPLoc2.0)对红螯螯虾DUF1943编码的蛋白进行亚细胞定位;NetPhos 2.0(http://www.cbs.dtu.dk/services/NetPhos-2.0)分析其磷酸化位点,借助SignalP 5.0在线服务器(http://www.cbs.dtu.dk/services/SignalP/),选择真核生物类型(Eukaryotes)进行蛋白常规的分泌通路(Sec/secretory)和I型信号肽酶(SPaseI),即真核生物仅含有的Sec/SPI信号肽进行预测。最后,采用在线工具SOPMA(https://npsa-prabi.ibcp.fr/cgi-bin/ npsa_automat.pl?page=npsa_sopma.html)分析蛋白的二级结构。
1.2.2 Pet28a(+) -重组表达载体的构建
根据序列的评估结果以及大肠杆菌的密码子偏好性,利用在线分析系统OPTIMIZER (http://genomes.urv.es/OPTIMIZER/")和DNA Works (https://hpcwebapps.cit.nih.gov/dnaworks/)对(617—918aa)序列进行密码子优化和序列改造,设计出目的片段的全基因合成方案委托生工生物工程(上海)股份有限公司合成。全基因合成的策略是将优化后的目的片段分割成42段带有重叠序列的寡核苷酸片段(即Oligos引物,其外侧首尾引物分别为:F: 5′-GACACAAATACTCTC-3′;R: 5′- GTGTCTTACGG-3′,下划线是H I和l的酶切位点),先将合成的42段寡核苷酸混合到一个体系中,通过第1轮PCR反应中多个热循环和退火实现目的片段的全长序列拼接,再利用首尾引物,通过第2轮PCR进一步扩增和富集全长目的基因。PCR反应采用的是高温聚合酶,第一轮重叠延伸PCR反应体系为50 µL:42对引物mix 0.5 µL×42,共22.0 µL(引物浓度为1 OD溶于400 μL ddH2O),dNTP(25 mmol·L-1)1.0 µL,10×Buffer 5.0 µL,0.4 µL(5 U·µL-1),补充ddH2O至50 µL。PCR反应程序为:95 ℃预变性3 min;95 ℃变性22 s,50 ℃退火20 s,72 ℃延伸30 s,20循环;最后72 ℃延伸5 min。以第1轮PCR产物为模板进行第2轮PCR扩增,反应体系为50 µL:首尾引物各2 µL(引物浓度同上),第1轮PCR产物1.0 µL,dNTP(25 mmol·L-1)1.0 µL,10×Buffer 5.0 µL,0.4 µL(5 U·µL-1),补充ddH2O至50 µL。PCR反应程序为:95 ℃预变性3 min;95 ℃变性22 s,55 ℃退火20 s,72 ℃延伸60 s,24个循环;最后72 ℃延伸5 min。对最后一轮PCR产物进行1.2%琼脂糖凝胶电泳检测并对特异性条带进行纯化回收。
选取pET28a作为表达载体,根据引物设计的酶切位点,利用H I和l对PCR反应后纯化回收得到的目的片段和质粒载体Pet28a(+)分别进行双酶切。酶切体系为50 µL:纯化的目的片段1.0 µg, 10×FD buffer 5.0 µL,H I 1.0 µL(10 U·µL-1),l 1.0 µL(10 U·µL-1), 补充ddH2O至50 µL。载体Pet28a(+)的酶切体系为50 µL:Pet28a(+) 1.0 µg, 10×FD buffer 5.0 µL,H I1.0 µL(10 U·µL-1),l 1.0 µL(10 U·µL-1)补充ddH2O 至50 µL。将上述两个体系分别放入37 ℃恒温水浴锅中反应2 h。对酶切产物分别进行电泳检测,并对目的产物进行纯化回收。
将酶切后纯化回收的目的基因片段和Pet28a(+)载体进行连接,连接体系为20 µL:目的片段的酶切产物8 µL,载体Pet28a(+)的酶切产物 4 µL,10×T4 DNA ligase buffer 2.0 µL,T4 DNAigase 1 µL (5 U·µL-1),补充ddH2O 至20 µL。将上述连接混合液放入16 ℃的PCR仪内反应1 h以上,转入Top10感受态细胞内,均匀涂布到含有卡拉霉素的LB平板上,37 ℃培养过夜,筛选出阳性克隆,提取质粒,再次进行酶切验证,反应体系为:质粒100 ng,10×FD buffer 1.0 µL,H I 1.0 µL(10 U·µL-1),l 1.0 µL(10 U·µL-1), 补充ddH2O 至10 µL,置于37 ℃恒温水浴锅中消化2 h。取5.0 µL消化产物进行电泳分析。经电泳分析正确后,将阳性克隆接种到含卡拉霉素的LB肉汤中,250 r·min-1,培养过夜,菌液送往北京华大(BGI)公司测序分析。测序采用的引物是Pet28a(+)载体上的T7启动子和终止子的通用引物序列(上游测序引物为:5′-TAATACGACTCACTATAGGG-3´,下游测序引物为:5´- GCTAGTTATTGCTCAGCGGG-3′)。
1.2.3 DUF1943蛋白的诱导表达 经电泳和测序验证后,将构建成功的重组质粒命名为 Pet28a(+)-,挑取阳性克隆转入到大肠杆菌感受态细胞BL21(λDE3),热激后涂布在含有卡拉霉素的LB平板上37 ℃培养过夜;挑取单克隆菌落接种到含有卡拉霉素的LB液体培养基中,37 ℃进行活化培养;当菌液OD600达到 0.6 时,添加一定浓度的诱导剂IPTG,分别于16 ℃以及20 ℃条件下培养16 h,37 ℃条件下培养4 h,设置未添加诱导剂的为阴性对照;取菌液经12 000 r·min-1离心5 min,弃上清,收集菌体沉淀;向其中加入PBS(pH 7.4)使其悬浮,利用超声破碎仪使其充分溶解,离心,弃上清,向沉淀中加入缓冲液A(8 mol·L-1Urea, 50 mmol·L-1Tris-HCl, 300 mmol·L-1NaCl, pH 8.0)进行溶解,再次离心,向上清和沉淀中分别加入等体积的2×Loading buffer煮沸,上样,进行10% SDS-PAGE电泳,由考马斯亮蓝(R250)染色的深浅程度来来确定蛋白表达情况,进而确定诱导表达所需的最佳的IPTG浓度、诱导温度以及诱导时间。最后,以最佳的诱导条件进行大量表达,菌液在4 ℃下经3 000 r·min-1离心10 min后,收集菌体沉淀。
1.2.4 DUF1943重组蛋白的纯化 向收集到的菌体中加入缓冲液B(8 mol·L-1Urea,50 mmol·L-1Tris,300 mmol·L-1NaCl,0.1% Triton X-100,pH 8.0) 4 ℃静置30 min,置于冰上进行200 W的多次间歇性超声破碎,见菌液澄清后,再行4 ℃、12 000 r·min-1离心10 min,收集上清粗蛋白(包涵体),加入2 mol·L-1的盐酸胍对其进行溶解,接着取 5 mL 的Ni-NTA柱子,用5倍柱床体积的 Binding buffer 清洗平衡柱子,流速 5 mL·min-1;将粗蛋白与平衡后的柱填料孵育1 h;将孵育后的产物上柱进行提纯和复性,收集流出;首先用Binding buffer 清洗平衡柱子;接着用Washing buffer 洗柱子,并收集流出液;最后用Elution buffer 再次洗脱,收集流出液,对粗蛋白、洗杂流出液、洗脱流出液分别处理,进行10% SDS-PAGE和R250染色分析。将纯度较好的6个组分分别加入到缓冲液C(50 mmol·L-1Tris,300 mmol·L-1NaCl,0.1% SKL,pH 8.5)进行透析,透析结束后用PEG20000浓缩,0.45 μm滤膜过滤后分装为1 mL·tube-1,–80 ℃保存。
1.2.5 DUF1943蛋白的鉴定 对复性且纯化后的目的蛋白进行制样,经SDS-PAGE电泳后,在200 mA恒定电流及低温条件下砖模(PVDF) 2 h,再用5% 脱脂奶粉的TBST封闭2 h后,将兔抗His-Tag单抗进行1 : 2 000稀释,HRP标记的羊抗兔IgG抗体进行1 : 5 000稀释,常温下孵育过夜,洗膜3次,将TMB显色液滴加到PVDF膜上进行显色,拍照分析,进行Western Blot特异性验证。最后,采用非干扰型蛋白定量试剂盒测定最终纯化产物的浓度。
2 结果与分析
2.1 DUF1943结构域的生物学信息
2.1.1 DUF1943结构域的基本理化性质 DUF1943结构域的的相对分子质量W为37 445.7,理论等电点pI为6.85,负电荷残基(Asp+Glu)总数为36,正电荷残基(Arg+Lys)总数为31,说明DUF1943结构域为带正电的蛋白。蛋白共计5 201个原子,分子式为: C1673H2562N454O498S14,不稳定系数为37.57,小于40,表明DUF1943结构域为稳定型蛋白。
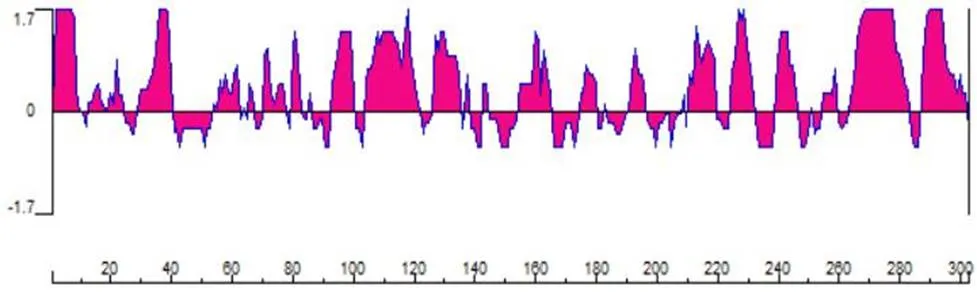
图1 DUF1943蛋白的疏水性分析
Figure 1 Analysis of the hydrophobicity of DUF1943 protein
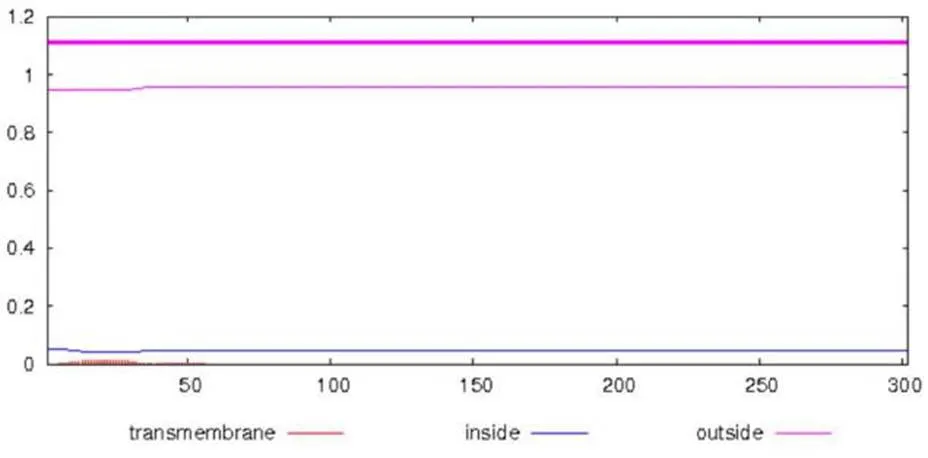
图2 DUF1943蛋白跨膜区预测分析
Figure 2 Prediction and analysis of transmembrane region of DUF1943 protein
2.1.2 DUF1943蛋白疏水性及跨膜区分析基因编码蛋白由336个氨基酸组成,其脂肪指数为76.37,亲水性平均值(grand average of hydropathicity, GRAVY)为- 0.285,结合Protscale的在线评估表明:序列中含有较多的亲水性氨基酸,亲水性区段主要为:1~9、11~22、29~40、52~61、70~82、91~99、102~121、122~132、151~163、172~180、190~193、210~220、222~231、238~245、255~260、262~282和289~302(图1),序列亲水性较好。疏水性氨基酸Ala、Cys、Ile、Leu、Met、Phe和Val分别占编码的1.80%、2.50%、4.50%、3.80%、1.90%、2.80%和4.20%。跨膜区分析结果显示(图2),DUF1943蛋白无典型的跨膜区,DUF1943蛋白的302个氨基酸均位于细胞膜表面,降低了跨膜区疏水性氨基酸对蛋白折叠的影响,易于表达和纯化。
2.1.3 DUF1943蛋白的抗原性分析 采用Protean模块中的的Jameson-Wolf方法结合预测DUF1943蛋白的抗原指数,发现DUF1943蛋白序列具有丰富且分布均匀的抗原表位位点,其中抗原指数较高的区段为:1~9、70~88、90~115、122~135、171~182、188~217、223~229、238~245、262~280和288~299(图3),并且这些区段具有强的表面可及性和亲水性位点,因此利用该蛋白制备抗体具有较强的可行性。
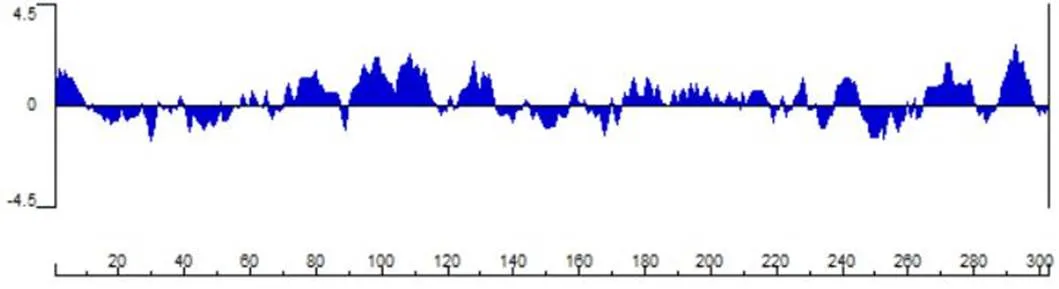
图3 DUF1943蛋白的抗原性分析
Figure 3 Antigenicity analysis of DUF1943 protein
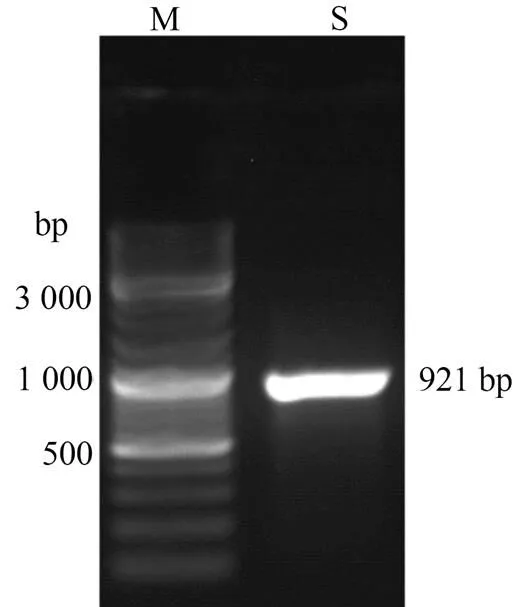
M: 10 kb DNA Marker; S: DUF1943基因的PCR产物。
Figure 4 PCR amplification ofgene
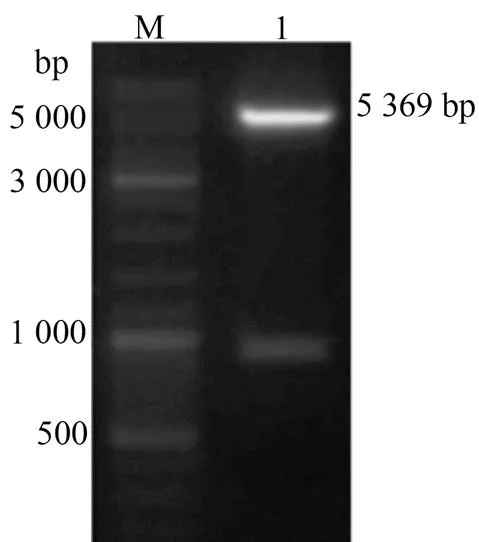
M: 10 kb DNA Marker; 1: pET-28a-双酶切产物。
图5重组质粒的双酶切鉴定
Figure 5 Double enzyme digestion identification of pET- 28a-
2.2 pET28a-DUF1943重组表达载体的鉴定
全基因合成所得的PCR产物经1.2 %的琼脂糖凝胶电泳检测显示在1 000 bp附近出现了特异性的目的条带,符合基因序列的预期大小(图4)。将合成的基因,连接到载体Pet28a(+)上,得到的重组质粒,经H I和l双酶切后,电泳检测结果显示如(图5)所示,酶切产物为两条大小约为5 300 bp和1 000 bp的片段,其条带大小与Pet28a(+)和基因的实际大小一致,初步断定目的基因已成功接入载体,Pet28a(+)-重组质粒构建成功。
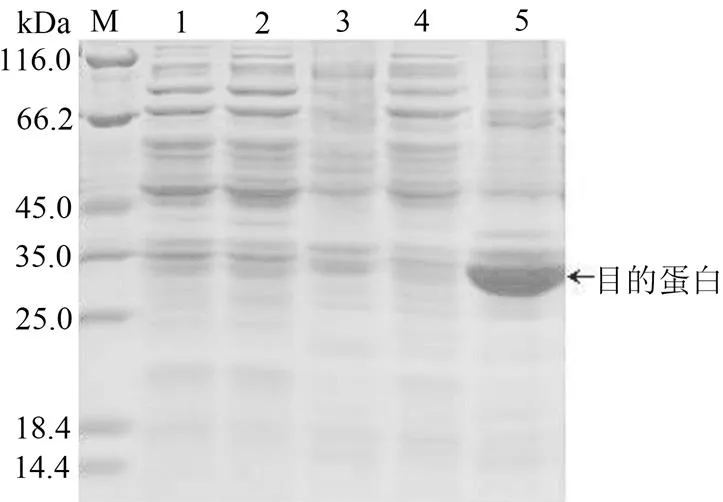
M:Protein Marker;1:诱导前总蛋白;2:20 ℃上清;3:20 ℃沉淀;4:37 ℃上清;5:37 ℃沉淀。
Figure 6 Expression of the DUF1943 protein in
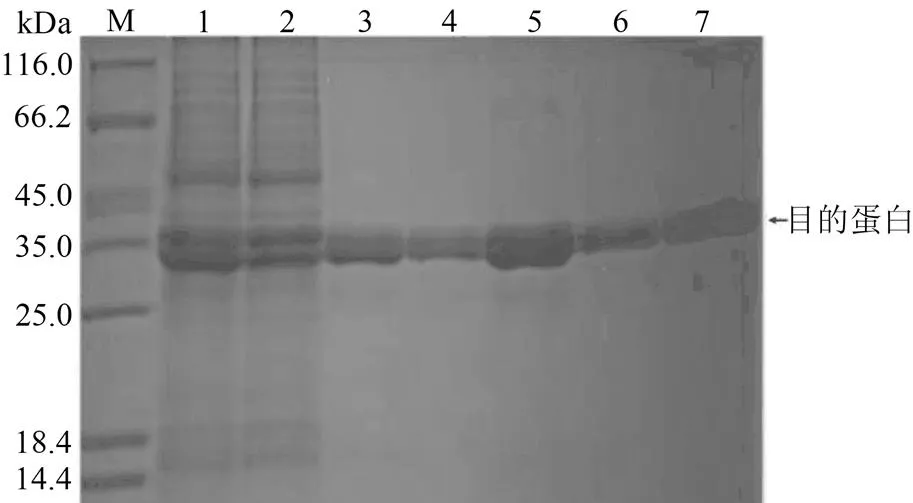
M: Protein Marker;1:上样;2:流出;3和4:20 mmol·L-1 Imidazole 洗脱组分;5和6:50 mmol·L-1 Imidazole 洗脱组分;7:500 mmol·L-1 Imidazole 洗脱组分。
Figure 7 SDS-PAGE analysis of the purified products by fusion protein nickel agarose affinity chromatography
对筛选出的阳性克隆进行增菌培育,菌液经测序分析,结果与NCBI上公布的红螯螯虾的Vitellogenin:AAG17936.1的相似性(identity)为100%,说明本次全基因合成并克隆转化所得的序列就是红螯螯虾Vg上的序列片段。可以确认Pet28a(+)-重组质粒构建成功。
2.3 DUF1943蛋白的表达鉴定
将重组质粒Pet28a(+)-转化大肠杆菌B21感受态细胞后,经IPTG诱导,进行SDS-PAGE电泳。结果(图6)显示,泳道5与其他泳道相比,出现了一条大小约35 kDa的特异性蛋白条带,大小与目的蛋白(37.4 kDa)预期一致。说明重组蛋白Pet28a(+)-在菌体内主要以包涵体的形式表达,且诱导其表达的最佳条件为:37 ℃培养4 h。
2.4 DUF1943蛋白的分离及纯化
将超声破菌、重悬后进行SDS-PAGE电泳检测,结果(图7)显示,各泳道在35 kDa附近都出现了一条明显的特异性条带,其大小与目的蛋白(37.4 kDa)一致。泳道3、4、5、6和7是采用不同浓度的洗脱液进一步纯化后的产物,与泳道1、2所示的未经洗脱的粗蛋白条带相比,条带更清晰,杂带更少。其中又以第5泳道的条带最清晰,纯度最高,说明50 mmol·L-1Imidazole为最佳的洗脱条件。
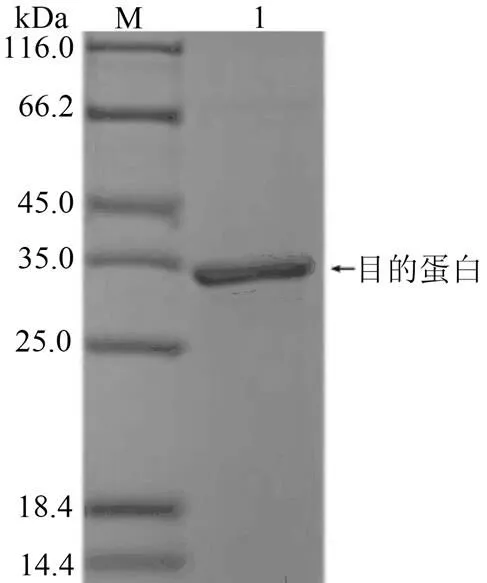
M: Protein Marker (Cat..No.C600525); 1: Pet28a(+)-纯化产物。
图8 纯化产物SDS-PAGE分析
Figure 8 SDS-PAGE analysis of the purified products

M:Protein Marker;1:融合目的蛋白。
Figure 9 Analysis of expressed recombinant DUF1943 by Western Blot
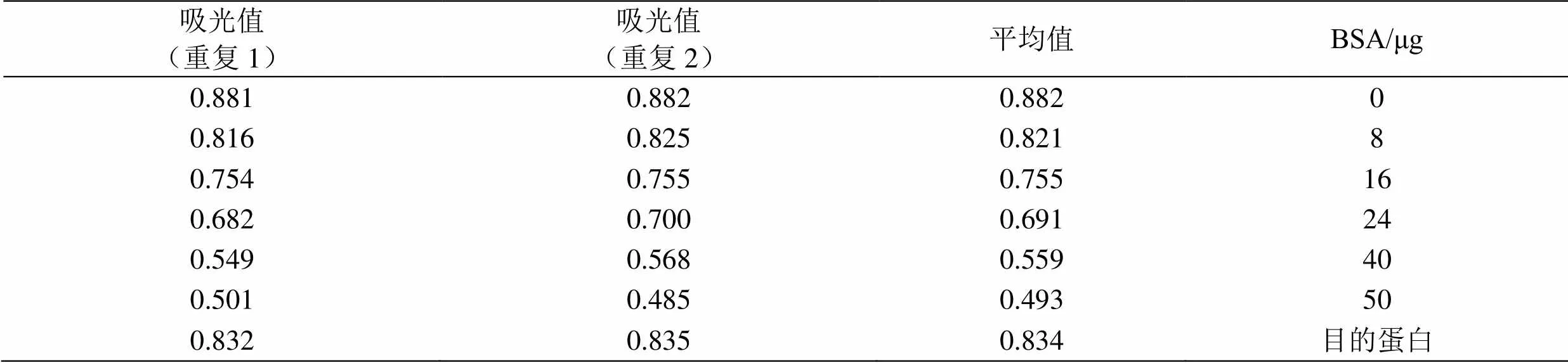
表1 Pet28a(+)-DUF1943蛋白浓度测定
注:待测样品体积为10 μL。
2.5 DUF1943蛋白的验证
收集最佳条件下大量表达的重组蛋白Pet28a(+)-,采用50 mmol·L-1的Imidazole洗脱液层析,得到的最终纯化产物经SDS-PAGE电泳分析。结果(图8)显示,在35 kDa附近出现一条清晰的目的条带,大小与预期结果一致。可确定纯化得到的即为目的蛋白。
为进一步验证最终纯化所得目的蛋白的特异性,对其进行Western Blot试验,并采用TMB进行显色,同样在35 kDa附近的相应位置出现了明显的特异性条带,再次表明该蛋白为目的蛋白(图9)。

图10 BSA标准曲线
Figure 10 The standard curve of BSA
2.6 DUF1943蛋白浓度鉴定
采用非干扰型蛋白定量试剂盒测定最终纯化蛋白的浓度,结果如表1和图10所示。将表1所测定的融合蛋白OD值代入标准曲线所得的回归方程,计算出融合蛋白Pet28a(+)-的浓度为0.60 mg·mL-1。
3 讨论与结论
研究发现卵黄蛋白原的DUF1943、DUF1944和VWD结构域都能够与革兰氏阳性和革兰氏阴性细菌结合,并能与其表面的病原相关分子模式LTA和LPS相结合[20]。Sun等研究发现斑马鱼的Vg2具有明显的抑菌作用,但是其DUF1943、DUF1944 和VWD结构域却不能够直接抑制细菌生长,而是作为模式识别受体与病原的相关分子模式相结合[21]。吴彪的研究发现虾夷扇贝的DUF1943能结合大肠杆菌和金黄色葡萄球菌及其表面的LTA和LPS,但是不能抑制细菌的生长,同样证明DUF1943作为模式识别受体发挥免疫学功能[22]。本研究先对红螯螯虾的进行了生信分析,重点评估了其编码肽段的抗原性进行了分析,发现其氨基酸序列区段具有较强的表面可及性以及较多的亲水性位点,表明DUF1943结构域可以作为一种高效的免疫原。而红螯螯虾DUF1943是否具有相应的免疫学作用,还需进一步的探研。制备DUF1943肽段正是为后续开展其相关功能及机制研究奠定基础。
用原核表达系统制备DUF1943结构域肽段的第一步,也是最关键的一步是目的基因的合成。重叠区扩增法(PCR-based accurate synthesis, PAS)仅需要2个步骤就能精准合成出在大肠杆菌宿主体内高效表达的目的基因,其错配率≤1/1 000 bp,适用于GC含量高,重复序列和二结构复杂的基因序列合成[23]。该方法首先通过对目的基因序列的理论评估,再根据大肠杆菌密码子的兼并性和偏好性,对序列中的大肠杆菌使用频率低的密码子和稀有密码子进行改造,使基因编码序列更符合大肠杆菌的密码子偏好。优化G+C的含量,添加或删除限制性内切酶位点,改变RNA二级结构,设计出优化的目的基因序列,对序列进行拆分,最终利用重叠区PCR扩增得到目的基因。高见等根据大肠杆菌偏爱密码子,利用Synthetic Gene Designer软件对L2E7全基因进行密码子优化,诱导其在大肠杆菌pET9a体内高效表达,目的蛋白占全菌的60%左右[24]。夏晓玲等根据大肠杆菌密码子偏好,利用GenScript rare codon analysis软件对十二指肠基因进行密码子优化,通过全基因合成方法合成的目的基因在大肠杆菌体内获得了较高的表达,且以可溶形式存在[25]。本研究先对红螯螯虾序列进行理论评估,根据大肠杆菌的密码子偏好,对红螯螯虾野生型序列进行的改造和优化,探索了诱导大肠杆菌表达DUF1943多肽的最优条件,最终获得了高效的DUF1943多肽以包涵体形式存在。包涵体经2 mol·L-1的盐酸胍溶解,Ni-NTA层析柱纯化及复性,最后透析去除变性剂,促进多肽折叠,恢复其活性。这是首次采用全基因合成方法对红螯螯虾Vg的DUF1943结构域进行高效的异源表达。
本研究利用全基因合成的方法合成了DUF1943结构域的片段序列,成功构建了Pet28a(+)-融合表达载体,探明了诱导大肠杆菌高效表达DUF1943多肽的最佳条件为:当单克隆菌液OD600达0.6时,添加0.6 mmol·L-1的诱导剂IPTG,置于37 ℃条件下培养4 h,蛋白的表达量最高。表达获得的DUF1943多肽以包涵体形式为主,经纯化和复性最终得到高纯度活性DUF1943多肽,为后续开展其具体生物学功能及实际应用奠定了基础。
[1] 蔡生力, 刘红.十足目甲壳动物卵黄蛋白原的生化特性及生物合成[J]. 水产学报, 2017, 41(1): 150-158.
[2] ABDU U, YEHEZKEL G, SAGI A. Oocyte development and polypeptide dynamics during ovarian maturation in the red-claw crayfish[J]. Invertebr Reprod Dev, 2000, 37(1): 75-83.
[3] OBERDÖRSTER E, RICE C D, IRWIN L K. Purification of vitellin from grass shrimp, generation of monoclonal antibodies, and validation for the detection of lipovitellin in Crustacea[J]. Comp Biochem Physiol C Toxicol Pharmacol, 2000, 127(2): 199-207.
[4] PAN M L, BELL W J, TELFER W H. Vitellogenic blood protein synthesis by insect fat body[J]. Science, 1969, 165(3891): 393-394.
[5] URIST M R, SCHJEIDE A O. The partition of calcium and protein in the blood of oviparous vertebrates during estrus[J]. J Gen Physiol, 1961, 44: 743-756.
[6] RIDGWAY U. A serological detected serum factor associated with maturity in English sole,, and Pacific halibut,[J]. Fish Bull - Natl Ocean Atmos Adm, 1967, 66: 47-58.
[7] BAILEY R E. The effect of estradiol on serum calcium, phosphorus, and protein of goldfish[J]. J Exp Zool, 1957, 136(3): 455-469.
[8] LASKOWSKI M. Über das Vorkommen des Serumvitellins im Blute der Wirbeltiere[J]. Biochem Zbl,1936, 284:318-321.
[9] VEERANA M, KUBERA A, NGERNSIRI L. Analysis of the vitellogenin gene of rice moth,stainton[J]. Arch Insect Biochem Physiol, 2014, 87(3): 126-147.
[10] ZHAO F, MORANDIN C, JIANG K, et al. Molecular evolution of bumble bee vitellogenin and vitellogenin-like genes[J]. Ecol Evol, 2021, 11(13): 8983-8992.
[11] MAZANKO M S, MAKARENKO M S, CHISTYAKOV V A, et al. Probiotic intake increases the expression of vitellogenin genes in laying hens[J]. Probiotics Antimicrob Proteins, 2019, 11(4): 1324-1329.
[12] KANG B J, JUNG J H, LEE J M, et al. Structural and expression analyses of two vitellogenin genes in the carp,[J]. Comp Biochem Physiol Part B: Biochem Mol Biol, 2007, 148(4): 445-453.
[13] XUE R, WANG X, XU S, et al. Expression profile and localization of vitellogenin mRNA and protein during ovarian development in turbot ()[J]. Comp Biochem Physiol B Biochem Mol Biol, 2018, 226: 53-63.
[14] FINN R N, FYHN H J. Requirement for amino acids in ontogeny of fish[J]. Aquac Res, 2010, 41(5): 684-716.
[15] SUN C, HU L L, LIU S S, et al. Functional analysis of domain of unknown function (DUF) 1943, DUF1944 and von Willebrand factor type D domain (VWD) in vitellogenin2 in zebrafish[J]. Dev Comp Immunol, 2013, 41(4): 469-476.
[16] SHEN Y, CHEN Y Z, LOU Y H, et al. Vitellogenin and vitellogenin-like genes in the brown planthopper[J]. Front Physiol, 2019, 10: 1181.
[17] WATTS M, PANKHURST N W, PRYCE A, et al. Vitellogenin isolation, purification and antigenic cross-reactivity in three teleost species[J]. Comp Biochem Physiol B Biochem Mol Biol, 2003, 134(3): 467-476.
[18] 杨树浩, 冯艺, 陈建酬, 等. 不同比例的生物饲料对罗氏沼虾生长、消化酶和免疫酶活性的影响[J]. 饲料工业, 2018, 39(2): 18-25.
[19] 罗文, 赵云龙, 姚俊杰, 等. 红螯螯虾胚胎发育期主要消化酶和同工酶的活性变化[J]. 动物学杂志, 2006, 41(1): 12-18.
[20] 李兆杰. 鱼类卵黄蛋白原的免疫功能研究[D]. 青岛: 中国海洋大学, 2009.
[21] SUN C, HU L L, LIU S S, et al. Functional analysis of domain of unknown function (DUF) 1943, DUF1944 and von Willebrand factor type D domain (VWD) in vitellogenin2 in zebrafish[J]. Dev Comp Immunol, 2013, 41(4): 469-476.
[22] 吴彪. 虾夷扇贝卵黄蛋白原基因克隆、表达和免疫功能研究[D]. 青岛: 中国海洋大学, 2014.
[23] 段桂华, 沈姗姗, 诸葛宇征. 改良重叠区扩增法构建Rac1相关质粒[J]. 生物技术通报, 2013(6): 177-182.
[24] 高见, 赵莉, 任皎, 等. 密码子优化的HPV16 L2E7基因在大肠杆菌中高效表达[J]. 中国医学科学院学报, 2007, 29(5): 579-583.
[25] 夏晓玲, 俞敏, 孙炜. 十二指肠钩虫ASP-2基因密码子优化及在大肠杆菌中高效表达[J]. 南京医科大学学报(自然科学版), 2014, 34(5): 627-633.
Prokaryotic expression and purification of the domain of unknown function of vitellogenin (DUF1943) in
WANG Rui1, WEI Zina1, LYU Min1, YANG Yanhao1, HUANG Liming1,WEI Xiuying2, LU Tianhe1, HUANG Guanghua1, YANG Huizan1
(1. Guangxi Academy of Fishery Sciences, Nanning 530021; 2. College of Animal Science and Technology, Guangxi University, Nanning 530005)
The aim of this study was to obtain the domain of unknown function of vitellogenin () fromby exogenous expression and then purification, so as to provide the material basis for further research on its related function and mechanism. According to thegene sequence (Accession No. AF306784.1) ofpublished in GenBank database, thegene ofwas found. The protein sequence of DUF1943 domain (617 - 918aa) was analyzed. The physico-chemical properties and basic structure of DUF1943 were evaluated theoretically. The codon ofwas modified and optimized according to the evaluation results, combined with the codon preference of. The optimized gene fragment ofdomain was obtained by whole gene synthesis method, and then it was connected into Pet28a (+) to construct the fusion expression vector of Pet28a(+)-. The optimal expression parameters of the fusion vector insystem were explored. The protein inclusion bodies were renatured by dissolution and upper column chromatography. The results of electrophoresis and sequencing analysis showed that the optimized target gene ofwas 921 bp, and the fusion expression vector ofwas constructed successfully. The optimal induction conditions for the high expression of the fusion vector inBL21 were as follows: when the OD600of monoclone reached 0.6, 0.5 mmol·L-1IPTG was added and cultured at 37 ℃ for 4 h; the expression of DUF1943 protein was mainly in the form of inclusion bodies, which showed a specific band near 35 kDa on SDS-PAGE. The inclusion bodies were broken and collected. After dissolution with 2 mol·L-1guanidine hydrochloride and Ni-NTA chromatography, the final concentration of active protein was 0.6 mg·mL-1. The prokaryotic expression vector Pet28a(+)-was successfully constructed, the expression system and renaturation scheme were optimized, and the target protein of DUF1943 was obtained, which would lay a foundation for the further study of the biological function and application of DUF1943 domain of Vg.
;; prokaryotic expression; purification; renaturation
S966.12; Q78
A
1672-352X (2021)06-0940-07
10.13610/j.cnki.1672-352x.20220106.021
2022-1-7 7:30:57
[URL] https://kns.cnki.net/kcms/detail/34.1162.S.20220106.1254.042.html
2021-02-01
广西科技重大专项课题(桂科AA17204095-5),广西自然科学基金项目(2019GXNSFAA185036),广西重点研发项目(桂科AB18221068)和广西科技重大专项(桂科AA17204094-7)共同资助。
王 瑞,博士,副研究员。E-mail:raywongxx@163.com
通信作者:杨慧赞,博士,高级工程师。E-mail:yhzyang@163.com
