Castleman病三例
2021-03-19聂金琳张浩然陈长顺姜海平
聂金琳 张浩然 陈长顺 姜海平
Castleman病(Castleman disease,CD)又称血管滤泡性淋巴组织增生症或巨大淋巴结增生症,由 Castleman等[1]在1956年第一次正式报道,是一种罕见的病因尚未明确的交界性非克隆性淋巴结增生性疾病,收录于国家卫生健康委员会等5个部门联合制订的《第一批罕见病目录》。CD不是真正意义上的肿瘤,常常无意间被发现,因其临床表现多样化并缺乏特异性,极易误诊、漏诊。我院2018年1月~2020年8月收治CD3例,我们对其临床特征及诊治过程进行分析,同时查阅并学习近年国内外与CD有关的文献以总结经验。
病例1,女,47岁。体检发现左后下纵隔阴影6个月余于2019年7月1日入院。外院多次胸部CT检查及胸椎MR检查报告均提示左后下纵隔旁(约胸11/12椎左旁)肿块,诊断为神经源性肿瘤或孤立性纤维瘤。入院后胸部CT检查提示左侧后下纵隔占位(图1a),诊断为神经源性肿瘤。AFP、CEA、糖类抗原均正常。行胸腔镜下左后下纵隔神经源性肿瘤切除术。术后病理诊断为透明血管型Castleman病(图1b)。术后经对症支持治疗后出院。
病例2,女,27岁。发现右颈部肿物9个月余,肿物进行性增大,伴体力下降、易激动、烦躁、多汗、饥饿、失眠于2019年5月5日入院。甲状腺彩超提示结节性甲状腺肿,右侧颈部多发淋巴结肿大。查体:右侧颈部可触及多个发肿大淋巴结,最大者直径约3 cm,质中,边界清,活动性可,无触痛,其余浅表淋巴结未触及肿大。入院后颈部MR提示右侧颈部囊实性病灶,诊断为甲状腺癌淋巴结转移或巨大淋巴结增生症(图2a)。甲状腺功能、CEA、AFP、糖类抗原均正常。行右颈部肿大淋巴结切除活检术。术后病理诊断为透明血管型Castleman病(图2b)。术后经对症支持治疗后出院。

图1a 脊柱左侧边界清晰的实性肿块影(箭头所示) 图1b 增生的淋巴样细胞呈“蚊香样”排列,包绕透明病变的小血管(HE,×20)
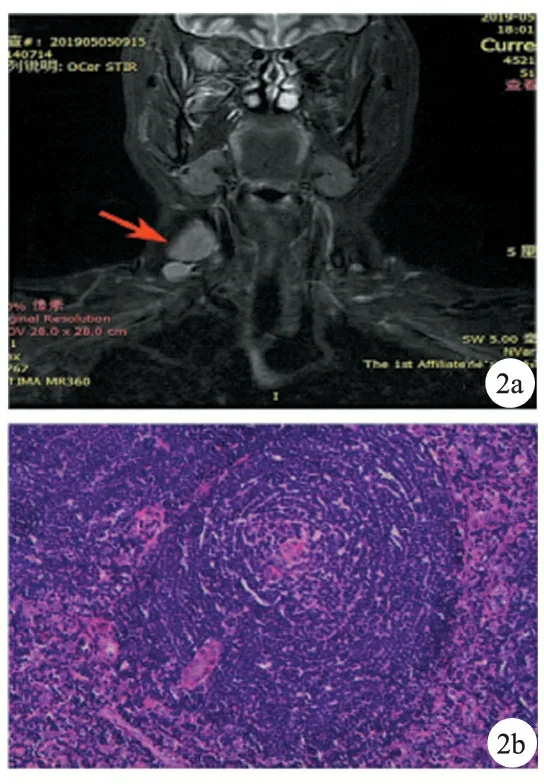
图2a 右颈部两个囊实性肿块影(箭头所示) 图2b 可见透明病变的小血管被“蚊香样”排列的小淋巴细胞包绕(HE,×20)
病例3,女,45岁。发现双侧腹股沟肿物进行性肿大13个月于2018年3月5日入院。查体:左侧腹股沟扪及直径约2 cm大小淋巴结,右侧腹股沟扪及直径约3 cm大小淋巴结,质中,边界清,活动性可,无触痛。AFP、CEA、CA19-9均正常。行双侧腹股沟淋巴结切除活检术,术后病理诊断为透明血管型Castleman病(图3a)。为了解肿大淋巴结分布情况行胸腹部CT检查,见弥漫多发淋巴结肿大,诊断为多中心型Castleman病(图3b)。病人拒绝治疗后出院。术后8个月,发现双侧腹股沟区淋巴结、双侧颈部淋巴结及左侧腋窝淋巴结进行性增大,伴腹痛、乏力。人疱疹病毒8(-),糖类抗原、CEA、AFP正常,骨髓涂片示浆细胞比例升高。予CHOP(环磷酰胺+阿霉素+长春新碱+ 泼尼松)+沙利度胺方案治疗一程,病人出现畏寒发热、全身乏力、腹胀、双下肢浮肿,更换R-CHOP(利妥昔单抗+环磷酰胺 +阿霉素 +长春新碱 +泼尼松)+沙利度胺方案治疗六程。治疗4个月后复查CT见各部位肿大淋巴结均明显减少、减小(图3c)。
讨论CD是一种罕见的病因未明的淋巴结增生性疾病,其发病原因可能与人疱疹病毒8感染、EB病毒感染、滤泡树突状细胞功能异常、免疫调节异常、血清白细胞介素水平升高、血管增生等有关系[2]。根据累及淋巴结的范围,临床上可分为单中心型(UCD)和多中心型(MCD)。根据病理学特点,病理学上可分为透明血管型(HV)、浆细胞型(PC)和混合型(Mix)。临床上以UCD多见,组织学多为HV。CD可发生在任何年龄段,好发于青壮年,男女发病率基本一致[3]。本组病例与文献报道相符的是:(1)发病率低;(2)无明确发病原因;(3)发病于青壮年阶段;(4)病理类型为HV。与文献报道不同的是本组病例均为女性病人。
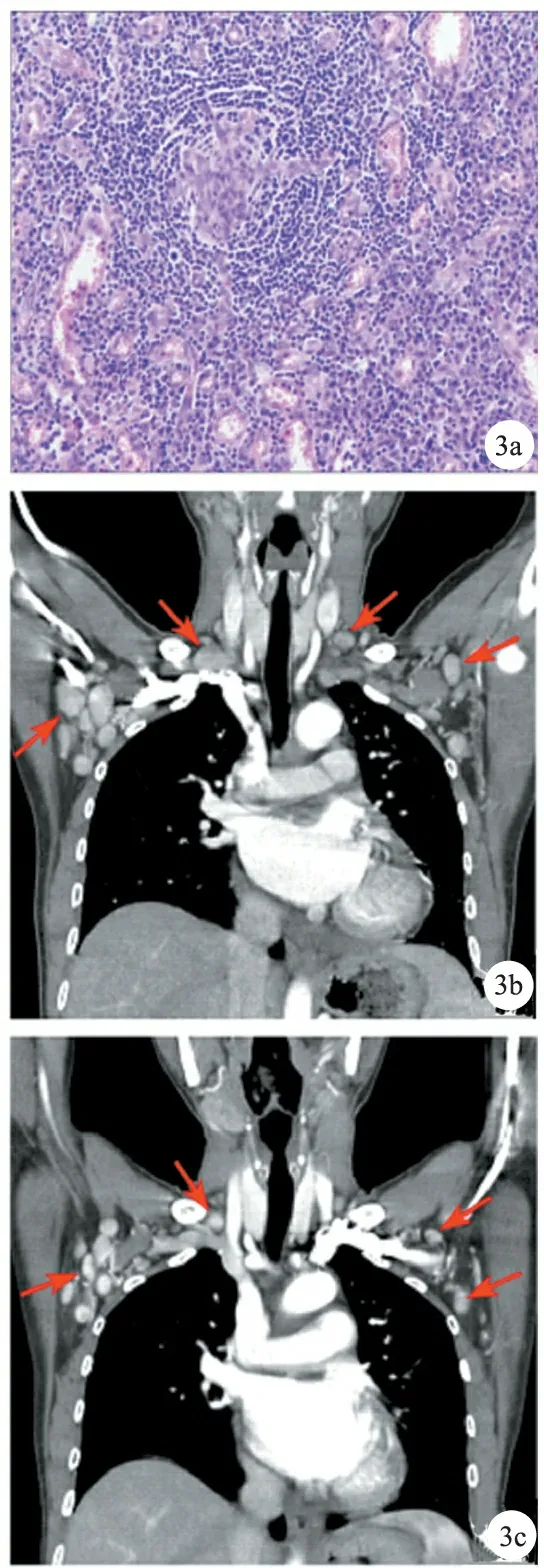
图3a 淋巴细胞呈“洋葱皮”样排列,包绕垂直插入的病变小血管形成“棒棒糖”样结构(HE,×20) 图3b 双侧锁骨上窝、腋窝大小不等的肿大淋巴结影(箭头所示) 图3c 双侧锁骨上窝、腋窝肿大淋巴结影较前减少、减小(箭头所示)
由于病灶的大小、部位及病理分型不同,临床表现多样性并缺乏特异性。常表现为淋巴组织样团块,可发生在任意部位的淋巴结,胸腔内尤其是纵隔最多见,也常见于颈、腋、腹部、肌肉及皮下等部位[4-6]。UCD通常表现为单一部位淋巴结的肿大,偶尔也会因为肿大的淋巴结压迫周围组织而出现相关的临床表现。而MCD除了全身多处淋巴结肿大以外,还可能出现贫血、乏力、发热、盗汗等全身性症状。MCD病人可同时合并POEMS综合征、干燥综合征、副肿瘤性天疱疮、系统性红斑狼疮等疾病[7],会出现与合并症的相关临床表现。本组病例均为无意间发现肿物,病灶部位各不相同,临床表现缺乏特异性。
UCD病人的实验室检查基本正常,而MCD病人常因相关合并症出现实验室检查异常。因此实验室检查对诊断多无价值。CD影像学通常表现为形态不一的软组织肿块影,难以与其他疾病相鉴别,极易误诊为神经源性肿瘤、淋巴瘤、淋巴结转移性肿瘤、间叶源性肿瘤、胸腺瘤、胃肠间质瘤等。因此,要确诊为CD,主要还需依靠良好的组织病理切片、免疫组化及基因检测等。由于穿刺活检术每次穿刺取材少,需要多次、多部位穿刺,但是影像学检查结果显示肿物富含血管,部分肿物出现在靠近心脏或大血管旁,反复穿刺活检风险极大,一般不建议行术前穿刺活检术,而是通过手术完整切除肿块送病理检查。本组病例实验室检查基本正常,在获得病理诊断之前仅1例考虑该病。
在治疗方案的选择、疗效及预后方面,UCD和 MCD有较大的差异[8-10]。UCD的治疗方案较为简单,一般以手术切除为主,预后好、治愈率高,复发率几乎为零。而MCD目前仍没有规范的治疗方法,因其具有潜在的侵袭性,预后不良甚至有可能发展为恶性肿瘤[11]。手术治疗主要用于减轻压迫或梗阻等症状,一般以联合化疗为主,CHOP和COP方案应用较为广泛,效果较为显著。免疫治疗、抗病毒治疗、靶向治疗和造血干细胞移植等方法也可取得较满意效果。对于MCD病人,合并症的治疗对预后也非常重要。本组2例UCD病人,尽管病灶部位不同,均予手术完整切除肿物,未予放疗、化疗等辅助治疗,术后分别随访13个月、15个月均未见复发。本组MCD病人因不能通过手术完整切除病灶,予联合化疗后病情得到缓解,随访期为29个月。
CD是一种罕见的病因和发生机制均未明确的疾病,其临床表现的多样化及异质性,导致其在诊断方面非常困难,误诊率极高,尤其是UCD,仅通过临床表现及体格检查往往难以诊断,影像检查对明确诊断的作用也很有限。因此,当诊断不明确时,病理检查可作为鉴别诊断的理想手段。到目前为止,国际国内尚无公认的、统一的诊断与治疗标准,CD诊疗方案的进一步完善,应引起广大临床工作者的重视。
