从药物多肽到蛋白质全合成:酶促拼接的方法原理与前沿应用
2021-03-18杨新宇朱彤李瑞峰吴边
杨新宇,朱彤,李瑞峰,吴边
(1 中国科学院微生物研究所,中国科学院微生物生理与代谢工程重点实验室,微生物资源前期开发国家重点实验室,北京 100101;2 中国科学院大学生命科学学院,北京 100049)
自20 世纪20 年代人类发现胰岛素以来,蛋白多肽类药物在体液调节、抗菌、抗炎、抗病毒及抗肿瘤等临床应用方面的重要性愈加显著[1-3]。截至2017年,全世界已有60多种多肽类药物获批上市,并以平均每年1种的速度持续增长[4-5]。虽然以重组表达为代表的现代分子生物学技术能够高效生产重组蛋白且可掺入个别非天然氨基酸,但难以合成含有多种非天然氨基酸的人工设计多肽与蛋白质[6-7]。目前多肽固相合成法仍是便捷获取非天然多肽的主要途径,然而该方法能够合成的多肽长度一般限于30~50个氨基酸残基,研究人员需要将多个多肽片段顺次拼接从而获得完整目标蛋白[8-9]。除此之外,许多待研究的多肽或蛋白质分子需要在特定位点连接寡糖链、脂类分子、核酸、荧光分子或另一个蛋白质分子等多样的功能基团。为了降低这些蛋白质的合成难度与成本,研究者采取了半合成策略,即利用化学方法合成一段带有功能基团的短肽,再将该片段与重组表达的蛋白质连为一体[10-11]。两段合成多肽的拼接以及蛋白质半合成均极具挑战,其难点在于需要保证酰胺缩合反应的区域选择性,同时必须抑制末端氨基酸残基的外消旋化副反应[12]。因此,探索具有严格区域选择性且尽可能避免外消旋的多肽拼接方法成为了蛋白质化学合成与修饰领域近年来的研究焦点。
现有的多肽拼接方法分为化学法与酶促法两大类。化学法包括自然化学连接(native chemical ligation,NCL)[13]、无痕施陶丁格连接(traceless Staudinger ligation)[14]、酮酸-羟胺连接(ketoacidhydroxylamine ligation,KAHA)[15]、丝氨酸/苏氨酸连接(serine/threonine ligation,STL)[16]以及二硒醚-硒酯连接(diselenide-selenoester ligation,DSL)[17]等(图1)。这些化学方法均采取了相似的策略,即两条多肽片段的末端化学基团发生选择性反应形成共价键,之后通过分子内重排形成肽键[12]。由Kent团队提出的NCL 是目前应用最为广泛的一种方法,该方法的原理为将一条多肽的C端活化为硫酯形式,而另一条多肽的N 端第一个残基固定为Cys,二者经过硫醇-硫酯交换与分子内重排两步反应形成肽键[18]。多肽硫酯可通过Boc 固相合成法(保护基为叔丁氧羰基)直接合成,也可通过Fmoc 固相合成法(保护基为9-芴甲氧羰基)制备多肽酰肼,再经过一步反应生成叠氮[19]或吡唑[20]中间产物,加入硫醇得到对应的多肽硫酯。若待连接片段为重组蛋白,则需要通过分子生物学方法在蛋白N 端添加Cys残基,或是将蛋白质C端活化为硫酯形式。目前最常用的EPL策略需在蛋白质C 端融合表达内含肽(Intein),之后在反应体系中加入苯硫酚以进攻内含肽与目标蛋白的结合处,从而形成硫酯[21]。NCL 方法的主要缺陷在于Cys是天然蛋白质中丰度最低的氨基酸种类之一,这严重限制了拼接位点的选择范围,尽管连接后可通过脱硫处理将Cys 变为Ala,但如果存在其他非连接位点的Cys残基,又需要增加保护与脱保护操作,这些步骤都会降低最终产物的收率[22]。此外EPL 策略最初使用的内含肽长约140 个氨基酸残基,虽然后续研究缩短了融合表达内含肽片段的长度[23],仍可能对重组蛋白的表达产生较大影响。
与化学法的原理不同,酶促法反应的区域选择性与立体选择性来源于酶活性中心的空间位阻以及基团间的非共价作用,满足多肽拼接在区域选择性及抑制外消旋方面的基本要求。目前研究较深的酶促多肽拼接策略主要有三种,分别使用Sortase A 转肽酶、Butelase 1 转肽酶以及Subtilisin人工连接酶,这些方法在拼接位点限制、蛋白质表达难度、酶活性等方面凸显出不同的优势,但在各个应用领域又分别有各自的限制,下文将系统阐述与比较。
1 多肽连接酶性质及其应用
1.1 Sortase A转肽酶
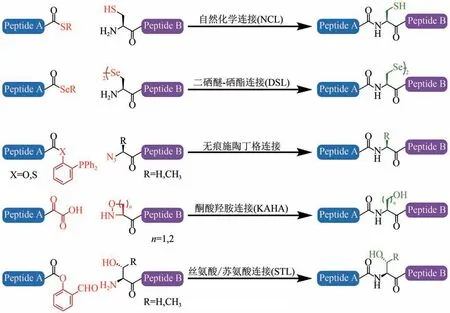
图1 主要化学连接方法[13-17]Fig.1 Chemical ligation methods[13-17]
SortaseA(SrtA)是源自革兰氏阳性菌的一种转肽酶,其中来源于金黄色葡萄球菌(Staphylococcus aureus)的SrtA 转肽酶应用最为广泛,该酶识别蛋白的LPXTG 序列(X 为任意氨基酸残基),切断苏氨酸和甘氨酸残基之间的肽键并形成酶-底物中间体,随后位于肽聚糖的寡聚甘氨酸肽桥进攻中间体,与目标蛋白C端之间形成新的肽键从而实现目标蛋白在细胞壁表面的锚定[24]。此外还存在识别LPXTA 序列的SrtA 转肽酶,这种来自化脓性链球菌(Streptococcus pyogenes)的转肽酶常出现在与金黄色葡萄球菌来源的SrtA 转肽酶联合使用的场合,两种酶对拼接位点的序列识别具有正交性[25]。SrtA 转肽酶可通过重组表达来大量制备,现已实现商品化,这让不同领域的研究者均能尝试以此为基础开发新型生物技术[26]。与内含肽介导的EPL 策略相比,使用SrtA 转肽酶催化蛋白质拼接时,仅需在目标蛋白末端融合表达长度仅有几个氨基酸残基的标签序列,虽然最终会留下一段“疤痕序列”[图2(a)],但与融合表达内含肽的策略相比,已大幅降低了蛋白质表达与折叠可能受到的影响,并且底物末端无需活化,获取底物的难度与成本较低。
2004 年,Mao[27]等首次利用SrtA 转肽酶进行蛋白修饰,在绿色荧光蛋白(green fluorescent protein,GFP)的C 端融合表达LPETG(H)6标签,再通过转肽反应将叶酸等小分子化合物或另一个GFP 分子连接至目标蛋白C 端[图2(b)],此后SrtA 转肽酶蛋白拼接技术很快被应用于多个生物学研究方向。细胞表面蛋白参与了细胞生长、分化、识别等众多生物学过程,一直作为重要的药物靶点受到广泛关注,Tanaka 等[28]将LPETGG 序列添加至重组破骨细胞分化因子C 端并运用SrtA转肽酶方法进行修饰[图2(c)],为细胞表面蛋白的时空动力学表征提供了新的研究思路,同样的方法亦可用于细胞表面蛋白的N 端修饰[29]。噬菌体表面展示技术是当下建立抗原抗体库、筛选药物等研究的常用手段,Hess 等[30]提出了组合应用衣壳蛋白的策略,通过在M13噬菌体衣壳蛋白pIII与pVIII 的N 末端分别添加寡聚Gly 和寡聚Ala(G5-pIII-A2-pVIII),先利用化脓性链球菌(Streptococcus pyogenes)来源的SrtA 转肽酶[Sortase A(strep)]将四甲基罗丹明(tetramethylrhodamine,TAMRA)修饰的七肽(KLPETAA)选择性连接至pVIII,再利用金黄色葡萄球菌(Staphylococcus aureus)来源的SrtA 转肽酶[Sortase A(staph)]将带有五肽标签(LPETG)的骆驼科重链抗体7(camelid heavy-chain antibody 7,VHH7)选择性连接至pIII,拓宽了噬菌体表面展示技术的功能范围[图2(d)]。与蛋白修饰相比,SrtA 转肽酶在环肽合成方面的成果较少,主要原因是连接后留下的“疤痕”较长,另外待环化多肽的长度一般需要超过19 个氨基酸残基,否则将倾向于肽段寡聚[31]。目前SrtA 转肽酶方法合成环蛋白的代表案例包括人唾液肽组蛋白Hst1[图2(e)][32]、重组胱氨酸结环肽rMCoTI-II[33]以及向日葵胰蛋白酶抑制剂SFTI-1[34]等,环化后蛋白的热稳定性以及抗蛋白酶水解能力均有不同程度的提升。
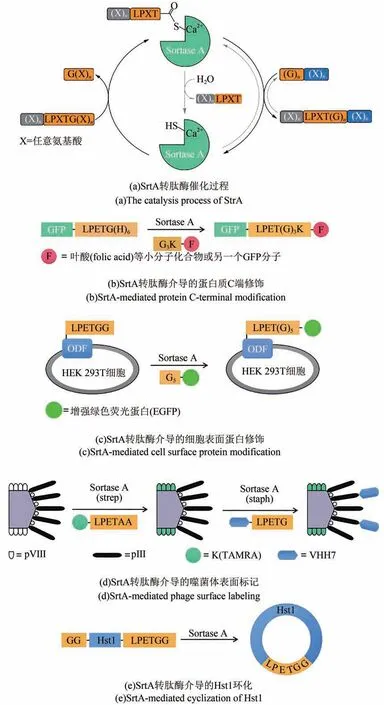
图2 SrtA转肽酶的催化过程及应用示意图[26-28,30,32]Fig.2 The catalysis process of Sortase A and its applications[26-28,30,32]
SrtA 转肽酶的主要不足之处在于其催化的连接反应是可逆的,因而需要较高的多肽底物浓度以保证连接效率,此外酶的催化速率偏慢,酶用量一般为蛋白底物物质的量的0.1~1倍[10,35-36]。当下研究者的一种优化思路是将SrtA 转肽酶与其他化学或酶学方法结合以取长补短,例如Muir 团队提出了基于内含肽自剪切技术与SrtA 转肽酶转肽反应的TAIL 策略,该策略可用于细胞核等复杂生物环境下的蛋白质末端不可逆连接,“疤痕”序列仅含一个Cys 残基[37]。清华大学刘磊团队[38]则用肼或肼衍生物替代寡聚甘氨酸底物进攻SrtA 转肽酶-底物中间体,产物变为不再被SrtA 转肽酶识别的蛋白质酰肼,由此可制备NCL 方法所需的蛋白质硫酯,或是在蛋白质末端引入炔基、叠氮等功能基团,通过点击化学反应(click reaction)完成二次修饰。SrtA 转肽酶的技术扩展案例为蛋白质合成与修饰领域指出了一个潜在的发展方向,即搭配使用现有的化学与酶促方法,建立充分发挥多种方法优势的多肽连接策略。
1.2 Butelase 1转肽酶
Butelase 1 转肽酶是热带药用植物蝶豆(Clitoria ternatea)合成环肽的过程中催化多肽环化的连接酶,识别多肽C 端的N/D-HV 序列并切断N/D 的C 端肽键形成酶-底物中间体,之后底物N 端进攻中间体从而完成多肽的环化[39-40]。Butelase 1 转肽酶的连接位点最终仅留下一个氨基酸残基(Asn或Asp)的“疤痕”[图3(a)],底物N 端的序列限制也比SrtA 转肽酶低得多,因此Butelase 1 转肽酶在环肽合成方面更具优势[40-41]。Tam 团队[42]利用Butelase 1 转肽酶合成了多种环蛋白,例如环状细菌素AS-48[图3(b)]、Uberolysin和Garvicin ML,其中AS-48 对包括李斯特菌(Listeria monocytogenes)在内的多种致病细菌具有优秀的抑制效果,其最小抑菌浓度低至0.1 μmol/L 数量级,为开发针对“超级细菌”的特效药物指明了新方向。可被Butelase 1 转肽酶环化的多肽长短不一,大到含有约250个氨基酸残基的GFP(环化速率约为SrtA转肽酶的20 000 倍),小到九肽,更短的多肽片段则一般会先寡聚后成环[43]。
实验表明,Butelase 1 转肽酶催化的多肽首尾环化反应不可逆,但两条多肽间的拼接反应是可逆的,释放的HV 二肽同样可以作为亲核试剂进攻酶-底物中间体,这导致Butelase 1转肽酶和SrtA转肽酶类似,需要较高的底物浓度来保证多肽拼接效率[44]。为了解决这一问题,Tam 团队[44]合成了C端序列为N-(thioglycolic)-V的多肽底物,此序列依旧能被Butelase 1 转肽酶识别,但释放的硫代二肽无法进攻酶-底物中间体,改进后的方法用于泛素N 端修饰时取得了高达95%的连接率[44]。不过在对连接率要求不是特别高的情况下,普通的底物已经能满足大部分需求,例如在大肠杆菌表面的OmpA 蛋白C 端添加NHV 序列,利用Butelase 1转肽酶即可将带有甘-异亮(GI)二肽的分子探针连接至OmpA 蛋白上,从而实现细菌活体标记[图3(c)][45]。当被修饰的蛋白质分子是一个以Lys为节点的多肽分支骨架时,就可以在这个“树干”上连接“枝叶”,将八条具有抗菌作用的四肽(RLYR)连接至这种骨架上[图3(d)],形成的树状多肽大分子的广谱抑菌活性相比单体有大幅提升,最小抑菌浓度降低了2 个数量级[46]。此外Butelase 1 转肽酶亦可用于蛋白硫酯的制备,目标蛋白C 端仅需额外表达NHV 三个氨基酸残基,再通过转肽反应连接多肽硫酯,无需内含肽的参与[图3(e)][47]。
Butelase 1 转肽酶的活性比SrtA 转肽酶高出2~3 个数量级,用酶量低至底物物质的量的百分之一,然而Butelase 1 转肽酶一直是从植物材料中提取,目前仍无法重组表达[10,48-49]。Tam 团队后续又发现了催化过程基本一致的OaAEP1转肽酶,该酶可在大肠杆菌中重组表达,但产量依旧较低(约2 mg/L),且kcat/Km值比Butelase 1转肽酶低2个数量级,经过酶工程改造后也仅能达到后者的三分之一[50]。获取困难成为了当下Butelase 1 转肽酶以及同家族连接酶推广过程的最大障碍,继续挖掘同家族的新酶是现阶段的一个重要研究方向。
1.3 Subtilisin 人工连接酶
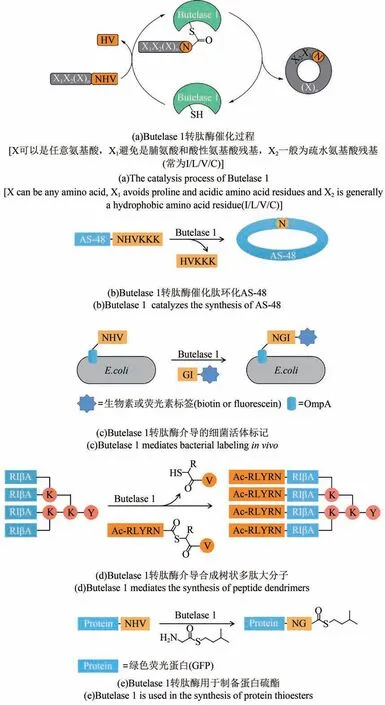
图3 Butelase 1转肽酶的催化过程及应用示意图[26,42,45-47]Fig.3 The catalysis process of Butelase 1 and its applications[26,42,45-47]
Subtilisin 是来自解淀粉芽孢杆菌(Bacillus amyloliquefaciens)的丝氨酸蛋白酶,具有六个广谱的氨基酸残基识别口袋,最初作为一种具有广泛切割位点的蛋白水解酶而受到关注。早在20 世纪60 年代,Bender 团队[51]即使用化学方法将Subtilisin 的关键活性基团从Ser 转变为Cys(S221C),得到的人工连接酶Thiolsubtilisin 在50% DMF 溶液中展现出了多肽连接酶的活性,但在水溶液中效率极为低下[52]。随着定点突变技术的发展,Wells团队[53]获得了重组表达的多肽连接酶Subtiligase,在S221C 之外还引入了P225A 突变来降低活性中心的空间位阻,将该酶的连接活性提高了1 个数量级且水解活性降低了2 个数量级,从此奠定了Subtilisin 人工连接酶的基本形态。之后Wells 团队[54]开展了一系列酶工程研究,先是额外引入5 个突变位点(M50F/N76D/N109S/K213R/N218S)以提高连接酶的稳定性,使得连接酶在4 mol/L 盐酸胍的环境中依旧能保留50%的活性;后续又在噬菌体表面展示技术[55]以及蛋白质组技术[56]的辅助下,改造得到一系列具有不同序列识别偏好的Subtiligase 突变体,从而扩展P1'与P2'口袋的底物谱。
相比SrtA 转肽酶和Butelase 1 转肽酶,Subtilisin 人工连接酶对连接位点的序列限制更少,不会出现“疤痕”序列[图4(a)],因此具有更广阔的应用范围。早在1994 年,Wells 团队[57]便利用Subtiligase 将6 个多肽片段(每段长度在11~31个残基)拼接为含有124个氨基酸残基的核糖核酸酶A,酰基供体多肽N 端被异烟碱(isonicotinyl,iNOC)基团保护以防止重复拼接,每轮连接反应后去除保护基来进行下一轮的连接[图4(b)],这比NCL 方法更早地实现了将多个多肽片段拼接为完整蛋白质[57]。Subtiligase还被用于环化长度在12~31 个氨基酸残基的多肽[图4(c)][58],在内含肽的辅助下可以实现蛋白质C 端的无痕拼接,例如Cole 等[59]利用Subtiligase催化重组表达的泛素硫酯与合成的十肽-生物素(GLSGRGKGGK-Biotin)的连接,解除了酰基受体肽N 端必须为半胱氨酸残基的限制[图4(d)]。不过Subtiligase目前最主要的应用方向仍是蛋白质N端修饰,Wells团队[60]建立了一个细胞凋亡相关蛋白水解酶研究平台,首先用Subtiligase 将带有生物素标签的多肽连接在混合蛋白样品的N 端[图4(e)],再用待研究的蛋白水解酶处理样品,酶切后用亲和素富集带有生物素标签的样品N 端片段,最后进行HPLC-MS/MS 分析[60]。生物素标记与亲和素富集的操作实现了酶解多肽片段的正向筛选,该平台可以帮助研究人员发现特定蛋白水解酶在细胞中的潜在靶标蛋白与切割位点。
除Subtiligase 之外,该家族的另一成员的研究也在近年获得突破。荷兰帝斯曼集团与格罗宁根大学合作,筛选了来自八十余种古菌和细菌的上百种Subtilisin 家族蛋白酶,确定了一个不依赖钙离子且高度稳定的模板蛋白,并对其活性中心进行改造设计,创制了一种新型多肽连接酶Peptiligase[4,61]。与Subtiligase 相比,Peptiligase 的折叠不依赖前体肽,表达量大幅提高且连接活性更高(酶用量可低至多肽底物物质的量的万分之三),还具有适用于工业生产的极高稳定性,足以耐受60 ℃的高温、高浓度DMF 与盐酸胍。后续帝斯曼集团将该部分业务独立,组建了全世界第一家应用生物法合成多肽与蛋白质的商业公司——Enzypep公司。通过对Peptiligase 的深入研究与改造,Enzypep公司推出了新一代广谱连接酶Omniligase-1,为低序列限制的通用多肽拼接策略提供了优秀的工具酶[62]。在多肽药物Exenatide 百克级合成过程中[图4(f)],Omniligase-1 催化下的两段多肽拼接效率达到88%,总产率是原先固相合成法单次合成完整多肽的两倍,展现了该酶极为突出的工业应用价值[63]。Omniligase-1 亦可与二硫键构筑[64]、点击化学[65]、肟连接(oxime ligation)[66]等方法联用以合成具有多环结构的环肽。不只是广谱连接酶,Enzypep 公司还推出了针对特定药物多肽序列的连接酶,例如专门用于生产Thymosin-α1的Thymoligase,该酶在P1 位特异识别带正电氨基酸,在P1'位特异识别带负电氨基酸,两段多肽底物的拼接效率高达94%,最终产率相比原有生产工艺提升了两倍[67]。Peptiligase 系列连接酶在连接/水解比、反应活性、底物序列限制等多个方面突破了工业化瓶颈,使多种药物多肽的生产成本下降了60%~80%,显著的工业应用优势让Peptiligase 系统被视为目前酶促多肽拼接领域最受瞩目的技术平台[68],针对不同应用方向来改造获得满足不同序列需求的突变体依旧是其未来的主要发展方向。
2 总结与展望
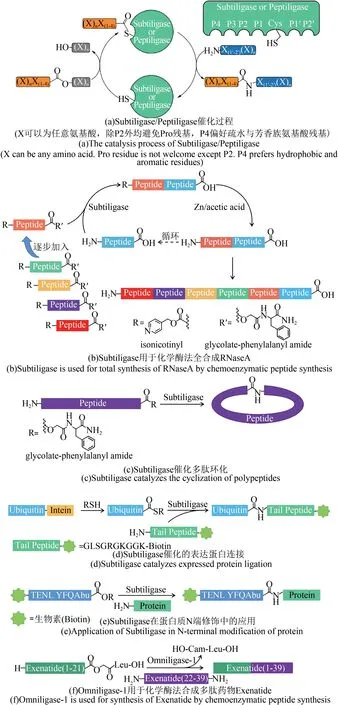
图4 Subtiligase/Peptiligase的催化过程及应用示意图[7,26,57-60,63]Fig.4 The catalysis process of Subtiligase/Peptiligase and its applications[7,26,57-60,63]
蛋白质重组表达与多肽固相合成技术在掺入非天然氨基酸、蛋白质表达系统适用性与多肽合成长度等方面都存在一定的限制,多肽连接技术可以将多个短肽连接成较长的肽段,同时解决了非天然氨基酸修饰与蛋白质合成长度的问题,而其中酶促多肽连接法由于其独特的区域选择性与立体选择性在蛋白质合成领域发挥着愈加重要的作用。近年来三种酶促多肽拼接策略经过优化与发展,各自具有其适合的应用场合(表1)。SrtA转肽酶可通过重组表达获得,也可以通过商业渠道购买,在蛋白质末端修饰方面便于使用,是目前应用案例最多的多肽连接酶,但在连接产物中会保留数个氨基酸残基的连接“疤痕”,不适合用于特定序列多肽或蛋白质的合成。Butelase 1 转肽酶活性较高,拼接位点的序列限制比SrtA 转肽酶少,连接后残留的“疤痕”较短,适合催化多肽与蛋白质的环化。然而该酶目前还无法通过重组表达制备,研究所用的酶需要从植物中获得,每千克的植物材料约能提取出5 mg 酶,难以实现大规模生产,这成为该酶应用的主要限制因素。Subtilisin 人工连接酶则在拼接位点序列限制方面优于前两种酶,只需要底物C末端活化为氧酯或硫酯而无需具备特定的氨基酸序列,虽然该酶的6个氨基酸残基识别口袋具有不同的氨基酸偏好性,但经过多年的研究优化,该系列酶已经产生了较多广谱突变体,并能整合具有互补选择性的连接酶突变体,建立的酶工具箱用于不同领域,功能最为全面[61]。并且Peptiligase系列商品酶是当下唯一能在工业级合成中使用的酶,不过这些商品酶大多价格高昂且序列保密,尚未普遍推广。三类酶促多肽拼接策略依旧具有较大的优化空间,研究者不仅可以联用化学和酶促方法来实现优势互补,建立不同的生物合成技术路线,还可以通过酶工程改造来提升连接酶的性能,过去三十年在这一方向已出现大量的理性设计与定向进化成果。而随着运算能力大幅提升以及先进算法不断涌现,近年来蛋白质计算设计得到了极大的发展,蛋白质从头设计的时代已经到来[69]。在蛋白质人工合成与蛋白质计算设计这两个领域相逢之际,计算机辅助手段既可以指导新型连接酶的设计与改造,提升连接/水解比,又能够帮助设计具有特殊修饰的非天然蛋白质[70],为酶促多肽拼接策略开拓崭新的应用场景。无论是化学方法与酶促策略的结合,还是人工合成与计算设计的相遇,均是不同领域的碰撞而闪现出的火花,深刻体现了学科交叉的意义与价值。相信在越来越多潜在相关领域的研究人员参与后,未来酶促多肽拼接策略的技术方法能再上一层台阶,应用于更多的多肽环化、蛋白质修饰乃至蛋白质全合成研究项目。
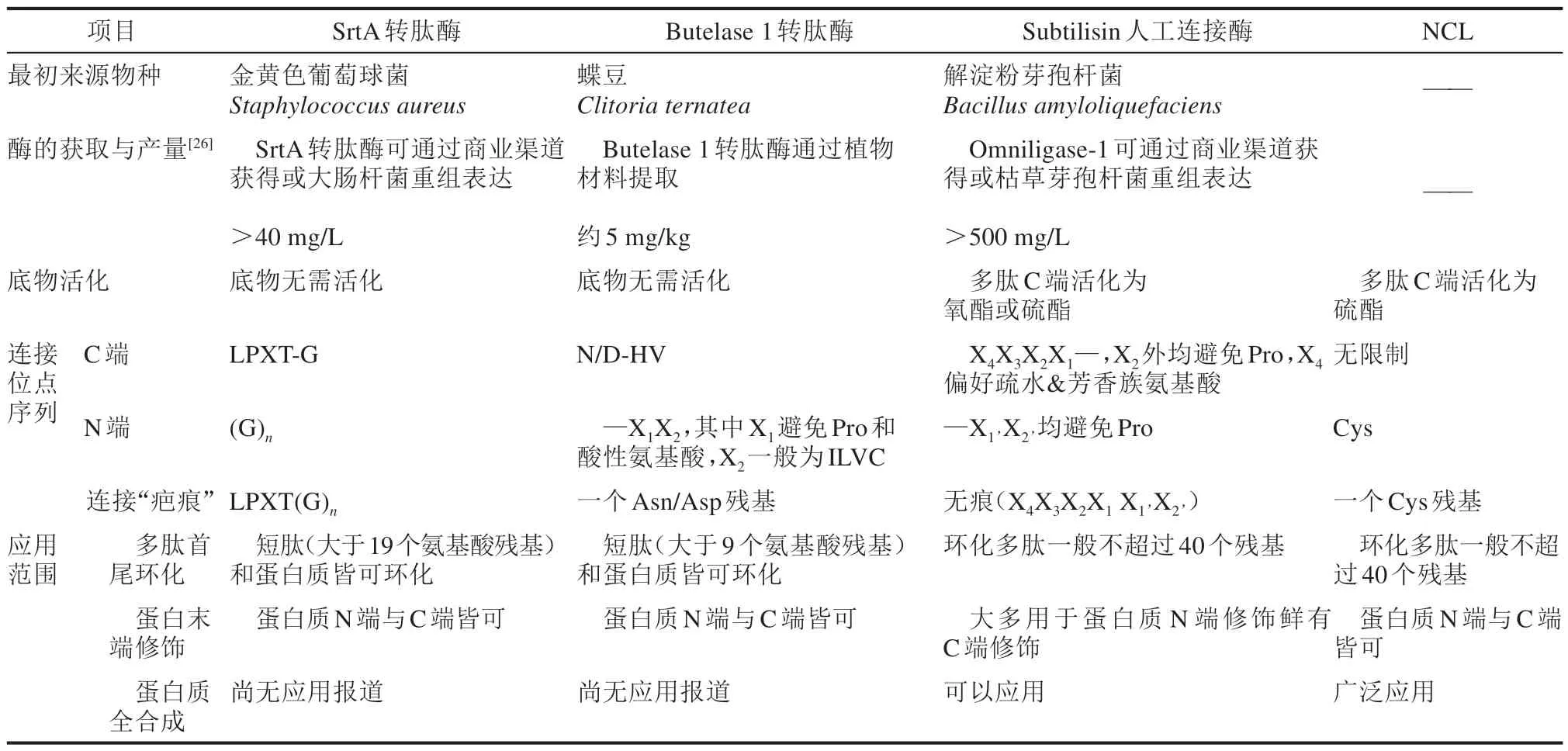
表1 三类酶促连接策略以及NCL方法的比较Tab.1 Comparison between NCL method and three enzymatic strategies
