老化对钴铈基催化剂催化氧化丙烷的影响
2021-03-17吴卫红刘少俊郑成航浙江大学能源清洁利用国家重点实验室国家环境保护燃煤大气污染控制工程技术中心浙江杭州3007浙江大学能源工程设计研究院有限公司浙江杭州3007
张 烁,吴卫红,杨 洋,刘少俊,宋 浩,郑成航,高 翔* (.浙江大学,能源清洁利用国家重点实验室,国家环境保护燃煤大气污染控制工程技术中心,浙江 杭州 3007;.浙江大学能源工程设计研究院有限公司,浙江 杭州 3007)
挥发性有机物(VOCs)被认为是臭氧、光化学烟雾和粉尘最常见的前体物之一,对环境和人体健康都构成了严重的危害[1-3].常见的VOCs,如烷烃、酮类和苯系物,大量来源于石化、焦化过程及发动机燃烧.随着排放控制法规的日趋严格,对低浓度VOCs的高效清洁技术提出了更高的要求.催化氧化法在较低的工作温度下具有较高的脱除性能,广泛应用于各种工业源治理[4].然而,丙烷等轻链烷烃结构稳定且热值较高,催化氧化的低温活性不足,正常运行后又容易出现催化剂床层局部过热的现象,一直是亟需解决的巨大挑战[5-6].
因此,催化剂需要兼顾良好的低温活性和热稳定性能.目前,贵金属基催化剂,特别是铂族金属催化剂被认为是低温下最具活性的催化剂[7-8].然而,贵金属催化剂成本高,活性受高温影响较大,亟需开发一种成本低、稳定性好、低温活性高的替代催化剂.在众多替代方案中,钴基催化剂由于优秀的催化性能,日益受到重视.研究指出,Co 氧化物呈现尖晶石状结构时,其活性远高于其他晶型的Co 氧化物[9],且尖晶石型氧化物独特的晶体结构使其不容易发生晶格的塌陷,是一种高稳定性的催化材料.Zhu 等[10]采用水热法制备了Co3O4/ZSM-5 催化剂,在230℃时氧化丙烷的转换频率(TOF)达到1.85s-1,并且在500℃保持40h 后仍未发现失活现象.尽管国内外对Co 氧化物的性质已有一定研究基础,但单独Co 氧化物的催化性能仍有待改善.
其中,Ce 以其良好的储氧性能闻名,是一种具有较高热稳定性能的助剂[11],有助于稳定活性组分结构,使其在混合氧化物体系中均匀的分散[12].此外,Ce4+的取代能够提升O2在催化剂表面的吸附能,有利于O—O 键的活化,从而提升催化剂低温的氧化活性[13].因此,将Co 良好的氧化还原性能与Ce 出色的储氧能力相结合,并保持尖晶石结构良好的热稳定性,是氧化丙烷催化剂的理想选择.然而,关于Co-Ce 复合氧化物催化剂可保持热稳定性能的温度,以及高温老化处理造成催化剂性能下降的主要因素仍有待研究.
本研究制备了Co-CeOx多活性中心催化剂,在不同的温度进行加速老化处理,选取较难降解的丙烷作为研究对象,测试了新鲜和老化后催化剂对丙烷的催化氧化性能,并进行了结构和性质的表征.通过扫描电镜、N2吸附/脱附和晶体结构表征,揭示了高温老化前后Co-CeOx催化剂的结构特征;并采用表面价态分析和程序升温还原实验,阐明了高温对催化剂表面化学性质的影响规律,构建了催化剂结构-性质-活性之间的关系,确定了Co-CeOx催化剂可以保持较高稳定性能的温度,揭示了高温老化处理影响Co-CeOx催化剂活性的主要因素,为氧化降解VOCs 的稳定运行提供了理论的参考.
1 实验方法
1.1 催化剂制备
将 0.045mol Co(NO3)3·6H2O 和 0.005mol Ce(NO3)3·6H2O 分别溶于25mL 去离子水中,将两种盐溶液均匀混合,加入柠檬酸使其与金属盐物质的量比为1.5:1,在水浴搅拌条件下保持80℃不断剧烈搅拌;当混合溶液变为深红色凝胶状时,将样品置于110℃的干燥烘箱中烘干12h,然后将样品移入马弗炉中450℃煅烧4h,最后经压片、研磨、后过筛后得到Co-CeOx催化剂样品.
加速老化处理的样品由Co-CeOx催化剂置于管式炉,在空气气氛下保持一定的温度煅烧24h,加速老化后的样品可标记为Co-CeOxT,其中T 代表了加速老化温度,依次为550,650 和750 ℃.
1.2 催化剂表征
采用美国Micromeritics 公司的ASAP2460 分析仪进行N2物理吸附-脱附实验,用来表征催化剂的比表面积和孔隙结构特性.X 射线晶体衍射(XRD)实验采用荷兰PANalytical B.V.公司的X'Pert PRO X射线衍射分析仪,以Cu Kα 为光源,在2θ=10~90°范围对样品进行扫描.
X 射线光电子能谱(XPS)分析在ESCALab220i-XL 型电子能谱仪上进行,用以获得催化剂表面元素的价态信息,分别得到了样品Co 2p 和O 1s 轨道的结合能,并以284.8eV 处的C 1s 谱图进行校准.
采用美国Micromeritics 公司的AutoChem II 2920 化学吸附仪进行H2程序升温还原(H2-TPR)实验.首先,称取50mg 催化剂样品,在Ar 气氛下保持350℃预处理1h,去除催化剂表面的杂质;接着,将催化剂样品降温至50℃,然后通入10% H2/Ar 的混合气,在该气氛下以 10℃/min 的升温速率升温至600℃,并用热导检测器(TCD)测量升温过程H2的消耗量.
1.3 催化剂性能测试
催化剂样品的活性评价在固定床反应器中进行,反应器为内径6mm 的石英管,采用800×10-6丙烷(根据实验需要换为丙酮或甲苯),20% O2和高纯N2模拟有机废气.使用质量流量计精确的控制气体的流量,保持反应过程中质量空速为90000mL/(g·h),采用芬兰Gasmet 便携式傅里叶变换红外烟气分析仪对反应器进口和出口处丙烷和CO2的浓度进行测量,记录丙烷转化率达到50%(T50)和90%(T90)的温度.丙烷转化率的计算公式如下:

式中:X 代表了丙烷的转化率,%;[C3H8]in和[C3H8]out分别为反应器进口和出口处丙烷气体的浓度,×10-6.
2 结果与讨论
2.1 催化剂活性测试结果
对新鲜的Co-CeOx样品以10℃/min 的升温速率分别在550,650 和750℃进行高温加速老化处理24h.对加速老化后的催化剂样品进行丙烷催化氧化活性评价,并与新鲜催化剂比较,其结果如图1(a)所示.新鲜的Co-CeOx样品催化氧化丙烷的T50和T90温度分别为213℃和248℃.经550℃加速老化24h后,催化剂的丙烷氧化效率与新鲜催化剂基本保持一致,T50和 T90温度略微升高分别为 216℃和253℃.650℃加速老化24h 后的催化剂样品氧化丙烷的T50温度为 225℃,较新鲜催化剂升高了约12℃,T90温度比新鲜催化剂提升了40℃.750℃加速老化后的样品在整个测试温度范围活性都有较大的下降,T50和T90温度分别上升了45 和125℃.因此,钴-铈催化剂在550℃以下的温度加速老化处理可以基本保持新鲜催化剂良好的氧化丙烷活性,老化温度继续升高会加剧对催化剂性能的影响,老化温度在650℃以上时尤为明显.

图1 Co-CeOx 样品高温加速老化前后丙烷氧化活性及CO2选择性Fig.1 Catalytic activity and CO2selectivity of propane oxidation over fresh and aged Co-CeOx catalysts
如图1(b)所示,新鲜催化剂在氧化丙烷过程表现出良好的CO2选择性,当温度高于225℃时,产物中对CO2的选择性均在95%以上.催化剂高温加速老化后,温度高于相应样品对丙烷氧化的T90温度时,应产物对CO2的选择性达到90%以上,但在低温段新鲜的催化剂CO2的选择性略优于高温加速老化催化剂,说明老化处理不仅影响催化剂的氧化反应活性,也一定程度影响了中间产物的深度氧化性能.

图2 Co-CeOx 样品催化氧化丙烷的耐久性Fig.2 Durability test results of catalytic propane oxidation over Co-CeOx catalysts
如图2 所示,新鲜催化剂在200℃时催化氧化丙烷的转化率在20%左右,稳定2.5h 后,升温至500℃保持约27h,Co-CeOx样品对丙烷的氧化效率一直维持在99%以上,没有活性下降的趋势.在30h 后重新降温至200℃,稳定后发现Co-CeOx样品催化氧化丙烷的效率仍然保持在20%左右.说明Co-CeOx样品在500℃具有良好的热稳定性,该过程基本对催化剂的活性不造成影响.
2.2 催化剂物理结构分析
通过ICP 表征,测算了不同催化剂样品中Co 的实际含量,如表1 所示,新鲜催化剂中Co 的摩尔含量为91.5%,与制备过程中设计的含量基本相同,确定成功合成了符合实验预期配方的催化剂.对新鲜催化剂和750℃加速老化后的催化剂样品进行表征分析,由图3 的XRD 图谱可以看出,新鲜的钴-铈催化剂表现出良好尖晶石结构的特征峰,未检测到单独的CeO2晶型特征峰,说明Ce 主要以无定形状态高度分散在尖晶石氧化物结构中[14].经高温加速老化处理后,催化剂的XRD 图谱衍射峰强度明显增强,并检测到较多的杂峰,在2θ=28.5°和46.8°处开始出现代表萤石结构的CeO2特征峰.随着老化温度的提升,萤石结构CeO2特征峰强度逐渐增强,这表明高温导致了催化剂部分晶型的转变,高温老化处理导致部分进入Co 的尖晶石结构中的Ce 物种析出,Co 和Ce 组分以单独的Co3O4和CeO2的形式存在,尖晶石状的混合氧化物结构部分被破坏.与老化处理的样品相比,新鲜催化剂的衍射峰明显较宽,采用Scherrer 公式计算750℃高温加速老化前后样品的晶粒尺寸,

式中:λ 为铜源的入射X 射线的波长,nm;θ 为X 射线相对于样品表面的入射角,°;β 为峰值半峰高处的宽度,nm;β0为仪器的谱线展宽,nm;K 为Scherrer 常数.结果如表1 所示,750 °C 高温加速老化后的样品平均晶粒尺寸显著增大,由21.4nm变为30.4nm,说明存在高温烧结的现象.较小的晶粒尺寸有利于提升活性位点的分散度,以及晶格缺陷的形成,使得氧化物暴露更多的活性晶面,从而提升催化剂的活性[15].因此,高温加速老化后催化剂丙烷氧化活性降低与晶粒尺寸增大有密切的关系.
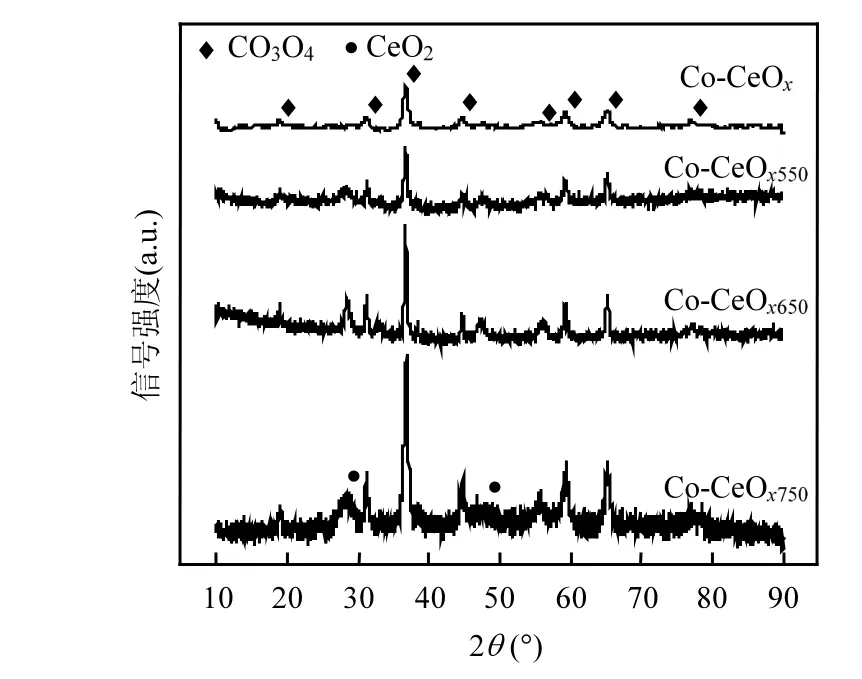
图3 Co-CeOx 催化剂高温加速老化前后的XRD 图谱Fig.3 The XRD patterns of fresh and aged Co-CeOx catalysts
高温加速老化前后催化剂的比表面积和孔道结构特征由N2吸附/脱附实验获得,结果如图4(a)所示.所有制备的催化剂均表现出具有H3 型滞后环的II 型等温吸脱附曲线,表明催化剂具有狭缝孔结构[16].新鲜催化剂样品的吸脱附等温线呈现更大的滞后环,说明具有更丰富的介孔结构,其BET 比表面积为65.63m2/g,孔尺寸为10.65nm;高温加速老化后,样品的比表面积和孔尺寸分别下降为60.15nm 和9.95nm.如图 4(b)所示,新鲜催化剂中孔径在20~50nm 之间分布最多,而老化后的样品10nm 以下的孔分布明显增大,这可能与晶粒长大及老化过程中介孔破坏有关.结合XRD 图谱的结果,高温的烧结现象使得晶粒长大,并与比表面积呈现负相关关系,这些因素都影响了催化剂对丙烷的氧化活性.

图4 高温加速老化前后Co-CeOx 催化剂的N2 吸附脱附和孔隙分布曲线Fig.4 N2 adsorption desorption curve and pore distribution curve of fresh and aged Co-CeOx catalysts

表1 高温加速老化前后Co-CeOx 催化剂的结构特性Table 1 Structural characteristics of fresh and aged Co-CeOx catalysts
如图5 所示,新鲜催化剂由非常细小的纳米颗粒构成,具有丰富的孔隙结构,非常的松散[17].高温加速老化处理后,550℃处理的样品未发生明显变化;随着老化温度的提升,颗粒逐渐团聚长大,650℃处理样品开始出现较多烧结塌陷产生的大孔;老化温度继续升高,孔道结构基本消失,750℃处理样品表面呈现较多烧结产生的半球形颗粒,颗粒尺寸显著增大.

图5 高温加速老化前后Co-CeOx 催化剂的SEM 图像(×50000)Fig.5 SEM images of fresh and aged Co-CeOx catalysts
2.3 催化剂氧化还原性能分析
如图6所示,新鲜催化剂中,在220℃附近的还原峰代表了—Co—O—Ce—结构中活性Co3+位点被还原为Co2+的过程,而300℃附近的还原峰表示了—Co—O—Co—结构中Co3+位点逐步被还原为Co2+的过程,400℃附近的还原峰表示了与Ce 弱相互作用的单独Co3O4中Co2+被还原为Co0的过程[18-19].此外,高分散活性Ce 物种的还原过程也发生在400℃附近[20-21].高温加速老化后,300℃附近Co3+的还原峰强度显著降低,400℃附近还原峰强度明显提升,这表明Co2+的比例较新鲜催化剂有所增大,与Ce弱相互作用的Co 物种的比例升高.这与XRD 的结果相照应,进一步反映了高温老化处理对Co-CeOx催化剂晶型的影响,使得Co 和Ce 之间相互作用减弱.然而,Co3+位点在丙烷的氧化反应中往往具有更高的氧化还原性能,Hu 等[22]研究指出Co3+-O3c位点上C-H 键的活化断键具有更低的能垒.Zhu 等[10]通过实验证明更高的Co3+比例和更强的Co3+氧化还原性能与丙烷的氧化活性密切相关,因此高温老化后丙烷氧化活性的降低可能与Co3+位点的减少有关,高温加速老化引发的催化剂结构变化进一步影响了催化剂的氧化还原能力.

图6 高温加速老化前后Co-CeOx 催化剂的H2-TPR 曲线Fig.6 The H2-TPR profiles of fresh and aged Co-CeOx catalysts
图7 为高温加速老化前后催化剂的XPS 图谱,其中Co 2p图谱如图7(a)所示,经去卷积分峰拟合后,可以看出在779,782,795 和797eV 附近存在四个子峰,并在788 和805eV 附近观测到卫星峰.以779 和795eV 为中心的峰对应于Co2+物种,以782 和797eV为中心的峰对应于催化剂表面的Co3+物种[23].Co 和Ce 之间的强相互作用使得电子由Co 向Ce 进行传输,从而导致催化剂表面产生了更多的Co3+物种[23],并形成了Co3+-O-Ce3+结构的活性位点.与新鲜的Co-CeOx催化剂相比,750℃高温加速老化后的样品中,Co3+/(Co2++Co3+)的比例由61.8 %下降为32.9%;高温下晶粒的长大使得Co3+物种难以在催化剂表面更充分的分散,同时CeO2的析出影响了Co 和Ce之间的相互作用,进而导致了催化剂性能的下降.O 1s 的XPS 图谱如图7(b)所示,位于531.1~531.5eV 的峰对应于O-、O2-和OH-等表面活性氧物种,可用Oα表示;在529.7~530.1eV 处的峰代表了晶格氧物种,可以用Ol表示[24].新鲜催化剂中,Oα/(Oα+Ol)的比例达到了49.6%,高温加速老化后Oα/(Oα+Ol)的比例下降至44.67%.研究表明,表面活性氧的含量与催化剂表面氧空位的数量呈现正相关的关系[25].催化剂表面的晶格氧从催化剂中解离,会在原来的位置形成氧空位,气体中的氧气分子比较容易被催化剂表面的氧空位活化.氧分子在氧空位处吸附,一个氧原子填补了氧空位,另一个氧原子成为表面活性氧物种,因此表面活性氧物种可以作为表面晶格氧与氧空位转化过程的指示物[26].因此,表面活性氧物种的流失,也可能是造成高温处理后的Co-CeOx催化剂活性下降的影响因素.此外,高温老化后,O 的XPS 峰位置向低结合能的方向移动,这表明催化剂中氧化物的形态发生改变,这与XRD 图谱和Co 2p 的XPS 图谱的结果相一致.图7(c)为Ce 3d 的XPS 图谱,通过去卷积分成8 个子峰,将Ce 3d5/2和Ce 3d3/2的自旋轨道分裂分别标记为u 和v,其中峰u'和v'对应于催化剂表面的Ce3+物种,峰u,u'',u''',v,v''和v'''归因于表面Ce4+[27].还原态的Ce3+存在表明部分Ce 进入了Co 的尖晶石结构中,形成了具有缺陷的复合氧化物结构[28-29].新鲜催化剂中,Ce3+/(Ce4++Ce3+)的比值为19.8%,高温加速老化后Ce3+的比例下降到了15.2%,进一步说明老化处理会导致部分进入Co 的尖晶石结构中的Ce 物种析出,Co 和Ce 组分之间的相互作用的减弱,不利于氧化反应过程中Co—O 键的活化和表面氧的解离,从而影响丙烷氧化的活性.

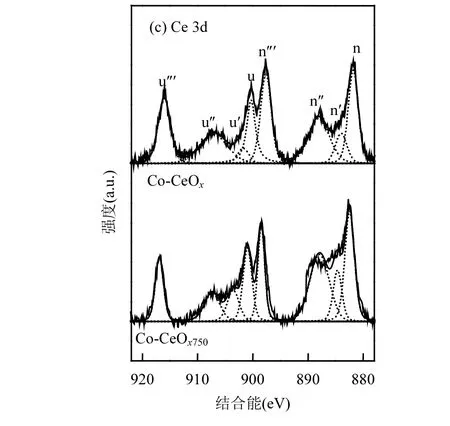
图7 高温加速老化前后Co-CeOx 催化剂的XPS 图谱Fig.7 XPS spectra of fresh and aged Co-CeOx catalysts
2.4 催化氧化不同VOCs 的反应性能
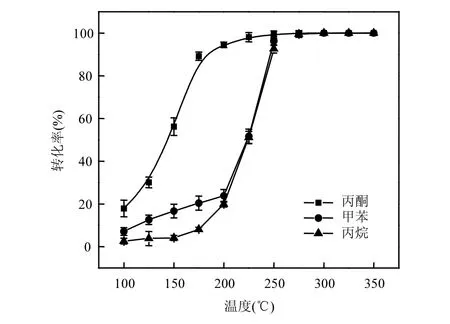
图8 Co-CeOx 催化剂氧化典型VOCs 的活性Fig.8 Catalytic activity of typical VOCs oxidation on Co-CeOx catalysts
进一步考察了Co-CeOx催化剂对丙酮、甲苯和丙烷等不同VOCs 的催化氧化性能,发现Co-CeOx催化剂对几种VOCs 都表现出了良好的催化活性,且反应物基本被完全氧化为CO2,未检测到CO 和其他副产物.Co-CeOx催化剂氧化丙酮、甲苯和丙烷的活性曲线如图8 所示.Co-CeOx催化剂氧化丙酮、甲苯和丙烷的T90温度分别为183,244和248℃.相对而言,丙烷由于碳链较短又只含有C—C 单键和C—H键,降解相对较为困难;而Co-CeOx催化剂上丙酮非常容易发生催化氧化,在150℃时转化效率已经接近60%;甲苯在Co-CeOx催化剂上的催化氧化开始较为缓慢,但在200~250℃之间活性迅速提升,也表现出优秀的催化氧化甲苯活性.因此,Co-CeOx多活性中心催化剂除了具有良好的丙烷氧化低温活性和热稳定性外,还具有一定的广谱性,对几种典型VOCs 污染物均具有良好的活性,表现出良好的应用前景.
3 结论
3.1 Co-CeOx多活性中心催化剂具有良好的丙烷氧化低温活性和热稳定性,T50和T90温度分别为210和235℃.当加速老化温度在550℃以下时,在测试温度范围内丙烷转化率变化幅度在3%以内;加速老化温度在650℃以上时,丙烷的转化率达到90%的温度开始明显升高.
3.2 催化剂高温加速老化处理后,比表面积减小,晶粒尺寸由21.4nm 变为30.4nm,物理结构的改变影响了活性位点的分散度,部分进入尖晶石结构的Ce 物种析出,Co 和Ce 的相互作用减弱,使Co3+/(Co2++Co3+)的比例降低至32.9%, Oα/(Oα+Ol)的比例下降为44.67%,氧迁移能力减弱,抑制了污染物在活性点位的吸附解离,导致了催化氧化活性的下降.
3.3 Co-CeOx催化剂氧化丙酮、甲苯和丙烷等典型VOCs 的T90温度分别为183,244 和248℃,在金属氧化物催化剂中具有一定的优势,表现出良好的应用前景.
