化学工程中机器学习的优势、限制、机会和挑战
2021-03-17MrtenDoelerePieterPlehiersRuenVndeVijverChristinStevensKevinVnGeem
Mrten R.Doelere , Pieter P.Plehiers , Ruen Vn de Vijver , Christin V.Stevens ,Kevin M.Vn Geem ,*
a Laboratory for Chemical Technology, Department of Materials, Textiles and Chemical Engineering, Ghent University, Ghent 9052, Belgium
b S ynBioC Research Group, Department of Green Chemistry and Technology, Faculty of Bioscience Engineering, Ghent University, Ghent 9000, Belgium
1.引言
在化学工程130年的发展中,数学建模对于工程师理解和设计化学过程而言非常宝贵。Octave Levenspiel甚至指出建模是化学工程中的主要发展[1]。如今世界快速发展,挑战比以往任何时候都要多。预测某些事件结果的能力是必要的,无论这些事件是否与新疾病活性药物成分的发现或合成有关,或者是否与为满足更严格的环境立法而提高工艺效率有关。这些事件的范围包括从表面反应的反应速率、反应器中反应的选择性到反应器中热量供应的控制。可以使用已经建立了几百年的理论模型进行预测。描述黏性流体行为的Navier-Stokes 方程[2‒3]就是这种理论模型的一个例子。然而,这些模型大多数都不能对现实系统进行分析求解,并且需要相当大的计算能力来进行数值求解。这一缺陷使大多数工程师首先选用简单的模型来描述现实情况。历史上,一个重要且对如今而言仍然相关的例子是普朗特边界层模型[4]。在计算化学中,科学家和工程师愿意为了缩短计算时间而放弃一些精度。与更高层次的理论模型相比,这种意愿解释了密度泛函理论的流行。然而,在许多情况下仍然需要更高的精度。
几十年的建模、模拟和实验为化学工程界提供了大量的数据,这些数据作为额外的建模工具包增加了根据经验进行预测的选择。机器学习模型是统计和数学模型,其可以从经验中“学习”,并在数据中发现模式,并且不需要显式的、基于规则的编程。作为一个研究领域,机器学习是人工智能(AI)研究领域下的子领域。人工智能是指机器执行任务的能力,这些任务通常与智能生物(如人类)的行为有关。如图1 所示,这并不是一个全新的领域。“人工智能”一词创造于1956年在美国达特茅斯学院为数学家举办的一个夏季研讨会上,该研讨会旨在开发更多具有认知能力的机器。从那时起,经过十数年的努力,人工智能技术才首次应用于化学工程中[5]。在20世纪80年代,更多的关注偏向于规则式专家系统,因为这被认为是人工智能最简单的形式。在那时,机器学习领域的研究已经开始兴起。但在化学工程领域,除去个别例外,机器学习的发展滞后了大约10年。20世纪90年代,随着聚类算法、遗传算法和最为成功的人工神经网络(ANN)的采用,关于人工智能在化学工程中应用的论文著述出版量突然增多。然而,这种趋势并非可持续的。Venkatasubra‐manian[6]认为这种兴趣的丧失可能是由于机器学习缺乏强大的计算能力和创建算法任务的困难性所致。
过去十年中,一个标志性的突破是深度学习的发展,深度学习是机器学习研究领域的子领域,它构建人工神经网络来模仿人类大脑。正如上文所提及的,人工神经网络从20 世纪90 年代开始在化学工程师中流行起来:然而,深度学习时代的不同之处在于,深度学习为多层神经网络的训练提供了计算手段,即所谓的深度神经网络。这些新发展激发了化学工程师的灵感,这从关于该主题的论文著述出版数量的指数级增长上也可以反映出来。过去,人工智能技术永远不会作为标准工具用于化学工程中;因此,对于当前是否是将之最终纳入标准工具箱的合适时机,是值得讨论的。本文将首先概述当今机器学习应用于化学工程的三个主要环节。本文接下来将批判性地讨论机器学习在化学工程中不断增长的潜力;文中将调查其利弊,并列出可能的原因来讨论为什么机器学习在化学工程中仍是“热门”的话题或为什么它最终会“不再热门”。
2.机器学习基础ABC
2.1.机器学习ABC中的“A”——数据
如图2所示,机器学习方法由三个重要环节组成:数据、表示和模型。机器学习方法的第一个环节是用来训练模型的数据。正如后面将要讨论的,所使用的数据也被证实是机器学习过程中最薄弱的环节。实际上,任何包含实验、第一性原理计算或复杂仿真模型结果的数据集都可以用来训练模型。然而,由于收集大量准确数据的成本很高,习惯上使用“大数据”的方法,即使用来自各种现有来源的大型数据库。由于真实实验的成本高昂,这些大量的数据通常是通过快速模拟或从专利和已发表的作品中进行文本挖掘获得的。数字化研究的增加为科学界提供了大量的公开资源和商业数据库。常用的化学信息来源有Reaxys [7]、SciFinder [8],用于反应化学和性质研究的ChemSpace[9],用于小的药物分子的GDB-17[10],以及美国国家标准与技术研究所(NIST)[11]和对溶解度等分子性质进行研究的国际纯粹与应用化学联合会(IUPAC)[12]。此外,还创建了几个基准数据集,以便在不同的机器学习模型之间进行比较。这些基准测试集的例子有用于量子化学性质的QM9 和Alchemy [13],以及用于溶解度的ESOL[14]和FreeSolv [15]。在使用任何数据集进行基于机器学习的建模之前,应该采取几个步骤来确保使用的数据质量足够高。确保数据质量的一般方面——从生成到存储——被称为数据管理。关于数据管理必要性和结果的更多细节将在下文进一步讨论。

图1.人工智能、机器学习和深度学习的发展时间表。关于人工智能在化学工程领域应用的出版物的发展表明,出版物数量上升之后是一个兴趣淡漠的阶段。目前,化工领域的人工智能研究再次处于“火热”阶段,然而现在尚不清楚曲线是否会很快回落。
机器学习(更具体地说是深度学习方法)与传统建模之间存在一些关于数据使用的差异。首先,人工神经网络从数据中学习并自我训练,而这样做需要大量的数据。因此,训练数据集通常包含数万到数十万个数据点。其次,数据集被分成三个而不是两个集:训练集、验证集和测试集。训练集和验证集都用于训练阶段,而只有训练集中的数据用于拟合。验证集是一个独立的数据集,为训练阶段提供对模型拟合的公正评估。测试集用不可见数据评估最终的模型拟合,并且通常是模型质量的主要指标。
2.2.机器学习ABC中的“B”——表示
机器学习方法的第二个重要环节是如何在模型中表示数据。即使数据已经是数字格式的,输入模型的变量或特征的选择也会对模型的结果产生重大影响。这一过程被称为特征选择,并且已经成为许多研究的热点话题[16‒19]。对所选择特征的数量进行限制可以减少训练和执行模型所需的计算成本,同时提高整体精度。这种特征选择过程在所谓的深度学习方法中相对不那么重要,因为深度学习方法被假定在内部已选择了那些被认为是重要的特征[20]。然后,一个由基本工艺参数(如压力、温度、停留时间等)、原料表征(如蒸馏曲线、原料组成等)或催化剂性能(如比表面积、煅烧时间等)组成的输入层通常是足够的[21‒27]。然而,在非数值数据(如分子和反应)的情况下,表征数据这一任务变得更具挑战性。
化学工程的任务通常涉及分子和(或)化学反应。为这些数据类型创建合适的数字化表征本身就是一个正在发展的领域。在计算机应用中,分子构成通常由基于线的标识符表示,如简化分子输入线性输入系统(SMILES)[28]或IUPAC国际化学标识符(InChIs)[29]或三维(3D)坐标。最近,自引用嵌入字符串(SELFIES)[30]是一种为机器学习应用设计开发的分子字符串表征。分子信息被转换成特征向量或张量,并输入到深度神经网络或其他机器学习模型中去。第一种表示分子的方法是选用一组分子描述符,如相对分子质量、偶极矩或介电常数[31‒33]。另一种生成分子特征向量的方法是从3D几何开始。基于几何表示的例子有库仑矩阵[34]、化学键分组(分子向量化表示)[35]以及距离、角度和二面角的直方图[36]。然而,在许多应用程序中,3D坐标或计算属性通常不可用。在这种情况下,可以从一个分子图开始创建表征,从而产生所谓的基于拓扑的表示方法。
基于拓扑的表示方法只可使用基于线的标识符。编码器可以使用自然语言处理技术直接将基于线的标识符转换为表示形式[37‒41],但通常是将基于线的标识符以类似于基于几何表征的方式转换为特征向量[42‒60]。这是通过向分子图中添加简单的原子和键的特征,然后在原子和键之间迭代传输信息来实现的。基于摩根算法[61]的圆形指纹[42‒46],如扩展连通性指纹[62],是机器学习应用的第一批分子表示形式之一。这些指纹就是所谓的固定分子表示,因为它们在机器学习模型的训练过程中不会发生改变。固定分子表示在药物设计中仍然流行,因为其可以快速预测候选药物的物理、化学和生物学特性[63]。由于在每个预测任务中,一个固定的表示向量代表一个分子,这种类型的输入层似乎与深度神经网络的定义相冲突,因为深度神经网络被假定为是从重要特征[64]中学习的。人们越来越倾向于学习如何表示一个分子[47,52],而不是聚焦在人类工程中的特征向量,因为人们认为,在数据更少、计算成本更低的情况下,更好地捕捉特征能确保更高的精度[53,58]。

图2.化学工程机器学习的三个主要环节;每个部分都对最终预测结果有影响,应该谨慎处理。
已学习的分子表征会被设计为预测模型的一部分。从几个初始的分子特征,如重原子、键类型和环特征开始创建分子表示方法,并且在训练期间进行更新。这种选择也表明,根据预测任务的不同,分子会有不同的表示方法。可以使用Gilmer等[59]综述的消息传递神经网络框架来描述广泛已学习的基于拓扑的表示方法[47‒58]。分子图中原子和键信息的加权转移是信息传递神经网络的特征。尽管有许多不同的表示形式存在,其复杂性各不相同,但值得注意的是,尚未开发出一种适用于所有类型分子性质的统一表示形式[65]。关于分子表示更详细的概述,读者可以参考David等[60]的综述。
就数据类型而言,化学反应比分子更为复杂。与基于线的分子标识符相似,化学反应可以通过反应SMILES[66]和反应InChI(RInChI)[67]来识别,而SMIRKS[66]可以识别反应机制。类似于分子,化学反应也应该被向量化以便在机器学习模型中发挥作用。最直接的方法是从反应物的分子描述符(如指纹)开始,对其求和[68]、相减[50,69],或进行串联[70‒72]。另一种方法是对于积极参与反应的原子和键,学习其反应表示[73]。反应也可以保存为文本(通常是InChI),通过神经机器翻译后,有机反应产物被视为反应产物的翻译[58,74‒78]。
2.3.机器学习ABC中的“C”——模型
机器学习方法的最后一个前提是建模策略。可供选择的机器学习模型种类很多。模型可以按不同的方式分类,可以根据其目的(分类或回归)或学习方法(无监督、有监督、主动或迁移学习)来分类。一般来说,术语“机器学习”可以应用于研究任何隐式建模数据集内相关性的方法[79,80]。因此,许多目前被称为机器学习方法的技术在被称为机器学习之前就已经开始使用了。其中两个例子是高斯混合建模和主成分分析(PCA),它们分别起源于19世纪下半叶[81]和20 世纪初[82‒83]。这两个例子现在都被认为是无监督机器学习算法。其他类似的无监督聚类方法有t分布随机邻域嵌入(t-SNE)[84]和基于密度的空间聚类(DBSCAN)在噪声场景下的应用[85]。图3 显示了监督学习和非监督学习技术之间的区别,并给出了非详尽的针对特定任务的有用算法的列表。在无监督学习中,算法不需要任何“解”或标签来学习;它会自己发现模式。无监督学习技术已经被用于化学工程的各种目的。Palko‐vits R 和Palkovits S[86]使用k-means 算法[87]根据催化剂的特征对其进行聚类,并使用t-SNE将催化剂的高维表示可视化。t-SNE不仅可用于催化,还是高维数据可视化的首选方法;它还被用于诊断化学过程的故障[88‒89]和预测反应条件[69,90]。主成分分析(PCA)是另一种降维算法,已多次被化学工程师用来确定训练集中占最大方差的特征[91‒97]。此外,PCA 还被用于异常值检测[93,98]。其他用于异常检测的算法包括DBSCAN 和长短期记忆(LSTM)[99‒100]。有兴趣的读者可以参考阅读Géron[101]的书以进一步了解机器学习算法。

图3.无监督和有监督机器学习算法的综述;非详尽列举了有用算法。ANN:人工神经网络;GMM:高斯混合建模;LSTM:长短期记忆。
当数据集被标记时,即已知每个数据点的正确分类时,可以使用如决策树(及其扩展方法随机森林)的监督分类方法[102‒103]。支持向量机是另一种可行的监督分类方法[104]。虽然支持向量机通常用于目的分类,但是也已经进行了扩展以允许通过支持向量机进行回归。回归问题需要使用有监督或主动学习方法,尽管原则上来说,任何有监督学习方法都可以归入主动学习方法中。人工神经网络(ANN)及其所有可能的变体[105‒113]是最常与机器学习联系在一起的方法。根据应用的不同,可以选择前馈神经网络(用于基于特征的分类或回归)、卷积神经网络(用于图像处理)或循环神经网络(用于异常检测)。化学工程师可能会遇到用于表示分子的卷积神经网络(见第2.2 节)[42‒60]、人工神经网络[32‒33,47,91,114‒117]、支持向量机[32]或用于预测表示性质的核岭回归[36,118]。人工神经网络已被作为黑箱建模工具应用于催化[23]、化工过程控制[119]和化工过程优化[120]等众多应用中。当已知标签时,对数据点进行分类的一种流行算法是k-最近邻算法,该算法已被用于化学过程监控[121‒122]和催化剂聚类[86,123‒124]。
3.优势
本节和接下来的几个小节将对化学工程师使用机器学习方法时的优势、限制、机会和挑战进行详尽的综述。图4概述了下面将描述的内容。
机器学习技术在化学和化学工程领域很受欢迎,因为它可以揭示人类科学家无法发现的数据模式。与明确依赖于物理方程(由已知模式推导出)的物理模型不同,机器学习模型并不只依赖编程来解决某个问题。对于分类问题,这意味着没有明确定义的决策函数必须被预先设计。对于回归问题,这意味着不需要推导或参数化详细的模型方程[80]。这些优点能有效地升级大型系统和数据集,而不需要耗费大量的计算资源。目前使用机器学习预测量子化学性质的热潮例证了机器学习技术的这些优点[32‒33,35‒37,39‒40,47,49‒50,52,55,65,68,71,73,115]。通常的从头计算方法往往需要花费数小时或数天来计算单个分子的性质,而训练好的机器学习模型可以在几分之一秒的时间内做出准确的预测。当然,其他能够准确预测的快速技术也已经开发出来了,但与机器学习模型相比,它们的应用范围有限[125]。机器学习的主要弱点是无法进行外推,但通过简单地添加新的数据点,可以很容易地扩展机器学习的应用范围。主动学习[126‒127]使得用最少的新数据扩展范围成为可能,这对于标记样本代价非常大的情况(如寻找数据点的真实值)是理想的,如量子化学计算[116]或化学实验[72,128‒129]。此外,现有的机器学习模型,如ChemProp[47]和SchNet[130‒131],可以随时使用,不需要经验。总的来说,机器学习在诸如scikit-learn [132]和TensorFlow [133]等软件包以及Keras [134](现在是TensorFlow [133]的一部分)或PyTorch [135]等框架下变得非常容易使用,这些框架将深度学习模型的训练限制在几行代码中。这样的软件包和框架使科学家有机会将他们的研究重点聚焦在研究的实际物理意义上,而不是把宝贵的时间花在开发高阶计算机模型上。

图4.在化学工程中使用机器学习作为建模工具的优势、限制、机会和挑战。
4.限制
机器学习方法的主要弱点之一是它们的黑箱本质。当给定某个输入时,机器学习方法将提供一个输出,如图5所示。基于模型在测试数据集上的统计性能,它可以对其输出的精度和可靠性做出某些陈述。模型超参数(如人工神经网络中的节点数)的详细分析可能是乏味的,但可以对模型已学习的相关性提供一些见解。然而,为某些行为提取物理上有意义的解释是不可行的。因此,无论其速度和精度如何,机器学习模型对于解释性研究而言不是一个很好的选择。
可解释性的缺乏增加了设计合适的机器学习模型的难度。与任何模型一样,机器学习模型会过拟合或欠拟合数据,而适当的模型位于两者之间。对于机器学习模型来说,过拟合的风险通常高于欠拟合的风险,这取决于训练数据的质量、数量和模型的复杂性。过拟合是模型结构的固有属性,并不依赖于超参数的实际值,这可以类比于用高阶多项式拟合(噪声)去拟合带噪声的线性数据集。在深度学习中,过拟合通常表现为过度训练,当模型多次显示相同的数据时,就会出现过度训练的现象。这导致模型记忆噪声而不是捕捉一般本质模式。通过将模型在训练数据上的性能与在验证和测试数据集上的性能进行比较,可以鉴别出过度训练。如果测试集的效果明显好于验证集的效果,那么模型可能训练过度。确定训练周期的数量往往很困难。为了避免过拟合,机器学习模型和其他优化问题一样需要一个停止准则。在传统建模中,模型通常涉及一些关于现实的至少某种形式的简化。由于包含简化,取得高精度的训练数据集是传统建模的主要挑战,所以这种停止准则通常基于训练数据集表现的变化而定。对于机器学习模型来说,实现训练数据集的精度通常不是问题;相反,挑战主要是当模型处于没有直接训练的情况下时,如何取得高精度的数据。因此,停止准则应该基于模型对“不可见”数据(即所谓的验证数据集)的表现而定。为了严格测试优化的数据集,需要一个完全独立的数据集——测试数据集,这也是传统建模方法中的常见做法。
机器学习方法的最后(但往往是最关键的)一个弱点是所使用的数据本身。如果数据集中存在过多的系统错误,网络本身也会产生系统错误,这就是所谓的“垃圾进—垃圾出”(GIGO)原则[136]。一些形式或来源的错误可以相对容易地被识别,而另一些错误一旦出现则很难被找到。如同每种统计方法一样,可能会出现异常值。相较于大的数据集,在小数据集上进行训练的模型更容易受到一些异常值的影响。这就是为什么在机器学习中不仅数据的质量很重要,数量也很重要。一种可能的解决系统性错误的方法是从数据集中手动删除这些数据点;也可以使用算法进行异常检测,如PCA[69,92]、t-SNE [137‒138]、DBSCAN[139‒140],或循环神经网络(长短期记忆网络)[111,141‒142]。近年来,基于自学习无监督神经网络的异常检测方法[143]已被开发出来[144‒146]。除了简单的异常值外,数据点也有可能是错误的。这种错误数据点可能来自一个测量错误实验中的样本,也可能来自一组不正确实验。例如,化学分析实验中仪器没有校准而产生的数据就是错误数据。在一组系统错误数据上进行训练尤其危险,因为模型会将错误趋势视为正确。通过对公开数据的认真审查,有可能发现上述问题。这个例子说明了数据管理的重要性,它确保所使用的数据是准确、可靠和可重复的。
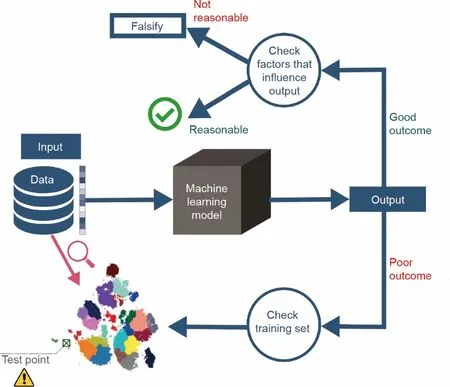
图5.解开黑箱模型的结果。不好的结果通常与所使用的训练集有关。当测试超出应用范围时,应发出警告信号。对好的结果需要加以验证,以了解模型学习到了什么。
显然,只有当数据可用时才能对其进行管理。尽管几十年的建模、模拟和实验为化学工程界提供了大量的数据,但这些数据通常存储于研究实验室或公司,因此不容易获得。即使可以访问数据,例如,读取内部数据库,获得的数据对机器学习而言可能也并非完全有用。使用文本挖掘技术从研究论文或专利中提取的数据[147]也存在同样的情况。这些数据可能没有用处的原因是一般情况下只会发表成功的实验,而失败的实验不会被发表出来[148]。此外,在人类化学工程师(具有洞察力和科学知识)看来毫无意义的实验或操作条件数据不会被执行。然而,机器学习算法却不具备这些知识,不包括这些“琐碎”的数据可能会导致预测错误。
5.机会
机器学习方法的许多优势提供了各种各样的应用机会,其最近的发展也缓和了一些针对机器学习的最重要的批评。几乎所有经过训练的机器学习方法都具有极高的执行速度,这使得这些方法非常适配于在预定义系统边界内需要精度和速度的应用程序。这类应用的例子包括前馈过程控制和高频实时优化[149‒151]。虽然对这些应用场景来说经验模型往往精度欠佳,但详细的本质模型却因难以快速运算而使得计算延迟无法被避免。基于本质模型训练的机器学习模型可以提供类似的精度,但需要付出经验模型的计算成本。在这种情况下,模型是基于高等级数据训练的,并试图预测经验结果和真实值之间的差异[152‒153]。无监督算法可用于过程控制应用以发现实时数据中的异常值[93]。机器学习方法是更准确、更快速预测与可靠的工业数据的结合,为创造数字孪生和更好的控制提供了机会,使得化学过程更为有效。
在多尺度建模方法中也可以得出类似的观察结果,在这种方法中,可以对各种不同尺度的现象进行建模,得到一组复杂且强耦合的方程组。机器学习在这类应用中的潜力很大程度上取决于多尺度方法的目标。如果目标是获得对低尺度现象的基本见解,那么机器学习就不可取,因为它具有黑箱特性。然而,如果将较小的尺度纳入该方法,以获得更精确的大尺度现象模型,那么机器学习可以用来替代较小尺度的缓慢的基本模型,而不影响大尺度现象的可解释性。
机器学习的最后一个机会在于解决其主要缺陷:不可解释性。可解释机器学习系统的问题并不是化学工程问题所独有的,它几乎存在于任何决策系统中[154‒157]。在催化领域,有人试图使机器模型所学习的内容可理解化[158]。然而,这种尝试仍然没有为模型结果提供任何层级的直白解释。图5显示了用于解释为什么会得到某个结果的工作流。当模型输出一个好的结果时,比如一个化学反应预测器给出了正确的产品,只有在检验了预测所凭借的基础之后,这个模型才应该是可信的。解释模型结果的第一步是量化个体预测的不确定性[159‒160],因为这提供了模型对其自身决策的置信度[115,161‒164]。一个相对简单的方法是通过集成建模。这种方法已经在天气预报中使用了几十年,并且可以与几乎任何类型的模型结合使用[165‒167]。人们还创建了一些算法来确定某些输入特征对输出的影响程度[168],或查看模型对某个输出使用了哪些训练点[169‒170]。当结果在化学或物理上看起来不合理时,应该寻找对抗性的例子来证伪模型而非验证模型[159]。而且,原因通常是在存在错误数据或偏差的数据集中发现的[171‒172]。
另一种使机器学习模型更具可解释性的方法是在模型中加入与化学相关且有充分根据的信息。虽然解释仍然需要大量的后续处理,但是如果使用人类可读的输入并且模型架构不是太复杂的话,这仍然是可行的。使用分子指纹作为输入的复杂递归神经网络几乎不可能被解释,因为人类很难破译这种模型输入。在风险管理中,经常采用“尽可能低的合理可行”(ALARP)原则[173]。类似地,为了让机器学习模型尽可能具有解释性,人们可以提出“尽可能简单合理”的原则。
6.挑战
机器学习模型的可访问性既是研究的主要优势,也是其主要挑战。虽然任何有基本编程技能的人都可以使用机器学习,但由于缺乏算法知识也可能导致误用。今天,有大量的机器学习算法可用,有可能有大量的参数和超参数组合。即使对有经验的用户来说,机器学习仍然是一种合乎逻辑的试错方法。由于研究人员经常无法解释为什么一种算法有效而另一种无效,一些人将机器学习视为一种现代“炼金术”[174]。此外,大多数已发表的文章不提供源代码,或仅提供伪代码,这使得研究人员不可能再现其算法[175‒176]。尽管机器学习在化学和化学工程领域不像社会科学那样面临许多可重复性问题[177],但由于该领域机器学习研究的增加,对其持怀疑态度的人可能也会相应增长。从Gartner 成熟度曲线[178]来看,机器学习和深度学习超过了膨胀预期的峰值[179],而且存在进入兴趣几乎消失的幻灭期的风险。除了不负责地任意使用算法之外,更危险的可能是对结果的错误解释。这种算法的黑箱特性使得很难甚至几乎不可能解释为什么会得到某种结果。此外,模型也可能因为错误的原因给出正确的结果[159]。因此,研究人员在使用机器学习时应该牢记统计学的一条重要规则:这是相关性的而非因果性的。
在应用超出模型所建立的范围时,就发生了另一种不合理地使用机器学习的情况。应用范围由训练数据集决定,并且是有限的。在测试未知数据点时,研究人员应检查这些数据点是否在应用范围内。当数据点超出范围时,用户应该会看到一个警告信号,提醒他们模型将表现不佳[92]。图5 的下半部分描述了如何通过查看训练集找到获得不当结果的原因。使用聚类算法的开源应用程序可以评估数据的精度及其应用范围[180]。
将机器学习应用于化学工程研究领域的最后一个挑战是,在机器学习技术方面,研究者受教育程度的差距越来越大。当在化学和化学工程中使用计算机和数据科学时,重要的是不仅要了解所使用的工具,还要了解其应用的过程。因此,在不久的将来,关于如何使用机器学习算法的简单培训可能会显得不足。相反,良好的人工智能和统计方法教育将在化学工程本科课程中变得至关重要。另外,在研究课题上,计算机科学家和化学专家之间需要更多的合作。训练不足的研究人员可能会错误地使用计算工具,而当计算机和数据专家不完全熟悉正在研究的主题时,他们可能无法得到最好的结果。更多的跨学科研究,以及机器学习专家和化学专家之间的合作关系,可能是避免对机器学习的兴趣进入幻灭期的一种方法。
7.结论和展望
在过去的十年里,机器学习已经成为化学工程师工具箱中的一个新工具。事实上,由于其具有执行速度快、灵活和用户友好的应用优势,化学工程师对机器学习的兴趣愈发浓厚。这种流行的另一面是误用机器学习或误解黑箱结果的风险,这可能会导致化学工程界对机器学习的不信任。以下三点建议可以帮助提高机器学习模型的可信度,使其成为一种更有价值、更可靠的建模方法。
第一,在化学工程界中保持对数据和模型简单、开放的访问非常重要。高质量的数据和开源模型鼓励研究人员将机器学习作为一种工具,使他们能够更专注于自己的主题,而不是花时间在编程和收集数据上。第二,且与第一点相关,是创建可解释模型。由于其他研究领域已经建立起机器学习,化学应用的新模型往往受到现有算法的启发。因此,研究为什么某个输出是由给定的输入生成的,而不是维护其黑箱特性,将有利于该领域的研究。第三条建议是对长远的算法教育进行投资。虽然化学工程师通常有很强的数学和建模技能,但理解图形界面背后的计算机科学是成为建模人员的前提。这也使定义模型的应用范围成为可能,这对于理解模型什么时候是插值,什么时候是外推至关重要。最后一点绝对是最为关键的:机器学习模型应该是可信的模型,这种可信度只有模型在多次训练集外的谨慎使用后方能获得。
Acknowledgements
The authors acknowledge funding from the European Research Council (ERC) under the European Union’s Hori‐zon 2020 research and innovation(818607).Pieter P.Plehiers and Ruben Van de Vijver acknowledge financial support, re‐spectively, from a doctoral (1150817N) and a postdoctoral(3E013419) fellowship from the Research Foundation—Flan‐ders(FWO).
Compliance with ethics guidelines
Maarten R.Dobbelaere,Pieter P.Plehiers,Ruben Van de Vijver, Christian V.Stevens, and Kevin M.Van Geem declare that they have no conflict of interest or financial conflicts to disclose.
