中年男性,记忆力下降2年,言语含糊、行动迟缓1年,头晕4天
——血浆铜蓝蛋白缺乏症☆
2021-03-17宋晓璇栾兴华曹立
宋晓璇 栾兴华 曹立
1 临床资料
患者,男,45岁,因“记忆力下降2年,言语含糊、行动迟缓1年,头晕4 d”就诊。患者2年来记忆力减退,如反复遗忘银行卡密码、时常丢东西,近1年明显加重,并伴有言语含糊、语速慢、行动迟缓及走路蹒跚。曾自主创业盈利,2年来逐渐无法胜任工作。4 d前无明显诱因出现头晕,伴有视物旋转、恶心,无头痛、耳鸣,无呕吐,2~3 min后自行缓解,共发作2次,间隔1天,每次发作后完全缓解,遂就诊于我院。发病以来饮食、睡眠、大小便尚可,近1年体质量下降约7 kg。
既往史和家族史:1型糖尿病史28年,常规胰岛素泵治疗,空腹血糖3~10 mmol/L。自幼学习成绩优异,大学本科学历。父亲房颤病史,母亲高血压病史,近亲结婚(患者祖母及外祖母系亲姐妹),妹妹体健,育有2子,均体健(图1A)。
体格检查:身高165 cm,体重63 kg,血压104 mmHg/62 mmHg。皮肤巩膜无黄染,浅表淋巴结未及肿大,甲状腺未触及,双肺呼吸音清,未及干湿罗音,心律齐,心率78次/min。腹平软,无压痛反跳痛,双下肢无水肿,双足背动脉搏动可。神经系统检查:神清,精神可,时间、地点、计算力、近事及远事记忆力粗测正常,简易智能精神状态量表(MMSE)22分。言语略含糊、缓慢,余脑神经(-),四肢肌张力轻度增高,肌力5级,共济稳准,闭目难立征(-),针刺觉对称,腱反射(++),病理征(-),脑膜刺激征(-)。
辅助检查:血红蛋白133 g/L(正常值120~160 g/L),平均红细胞体积 82.2 fL(正常值83.9~99.1 fL),尿葡萄糖(++++),糖化血红蛋白7.6%,糖化血清白蛋白21.19%,GADAb 2.06 U/mL(正常值0~5 U/mL)、IAA阴性、ICA阴性。铁代谢:铁蛋白1095.2 ng/mL(正常值80~130 ng/mL),血清铁 4.1 μmol/L(正常值9~29 μmol/L),铁饱和度 8.9%(正常值20%~55%),总铁结合力45.9 μmol/L(正常值45~72 μmol/L),转铁蛋白236 mg/dL(正常值220~400 mg/dL)。铜蓝蛋白<2.00 mg/dL(正常值20~40 mg/dL)。肝、肾功能、血脂、电解质、肿瘤标记物,叶酸、维生素B12未见明显异常。头颅MR平扫:双侧基底节区灰质核团对称性短T2信号,伴双侧壳核及尾状核尾部信号异常(图1B)。腹部MR平扫:肝脏、胰腺重度铁质沉积(图1B);脂肪肝,胰腺脂肪浸润;胰腺轻度萎缩。脑电图:额颞区较多段状慢波活动。神经电生理检查正常。眼科检查:糖尿病I期眼底病变,无K-F环,未见视网膜变性。
基因检测:患者于铜蓝蛋白基因(Ceruloplasmin,CP)检出一个纯合突变c.C643T,该突变位于第4号外显子,可致第215位精氨酸转变为终止子,该位点于ExAC及1000 g人群数据库及本地数据库均未收录,依据ACMG分级指南可评估为致病。Sanger测序证实先证者于该位点存在纯合变异(图1C)。该突变最早于2013及2019年收录于clinvar数据库[1],目前尚无该位点的功能学研究报告。
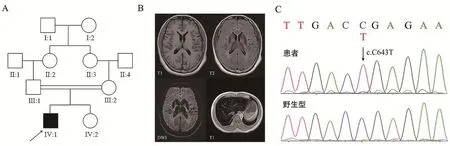
图1 患者家系图、影像学检测及突变分析(A)箭头所示为先证者。影像学检测(B)示双侧基底节区灰质核团对称性短T2信号灶,伴双侧壳核及尾状核尾部信号异常,肝脏、胰腺重度铁质沉积。(C)先证者于CP基因存在纯合突变c.C643T(p.R215X)。
2 讨论
本例患者以青少年起病的胰岛素依赖型糖尿病为首发症状,中年出现中枢神经系统症状如记忆力减退、言语含糊以及运动迟缓。MRI提示肝脏、胰腺以及基底节显著铁沉积,血浆铜蓝蛋白极低,伴铁蛋白增高。CP基因出现c.C643T纯合突变,提前形成终止密码子致使铜蓝蛋白提前截短,进一步证实该患者为CP基因突变所致的血浆铜蓝蛋白缺乏症。
血浆铜蓝蛋白缺乏症(aceruloplasminemia,ACP)是一种罕见的常染色体隐性遗传性脑组织铁沉积性神经变性疾病(neurodegeneration with brain iron accumulation,NBIA)[2-3],由CP基因突变所致,可直接影响体内铁代谢内稳态。ACP患者肝脏、胰腺和中枢神经系统等多系统可存受累[3],其中约68.5%的患者以胰腺铁沉积所致胰岛素依赖型糖尿病为首发症状,平均38.5岁起病,90%患者后期可出现诸如认知功能障碍、共济失调、不自主运动及帕金森样症状等中枢神经系统症状,80%~87%的患者有小细胞或正细胞性贫血[4-5]。尽管铁沉积和感光细胞的丢失会引发视网膜变性,且在半数以上的患者中出现[6],但极少作为首发症状就诊,故Langendonk提出将糖尿病、贫血和神经系统症状作为血浆铜蓝蛋白三联征,取代先前糖尿病、视网膜变性和神经系统症状的三主征[4]。神经系统症状中,约20%的患者伴有帕金森样表现,如肢体僵硬、运动迟缓等,本例中观察到的患者行走迟缓,可能是帕金森样症状的体现[7]。而金属储存障碍,如铁沉积,是帕金森样症状的重要原因[8]。ACP患者脑病理检查可见神经元与星形胶质细胞中均出现铁沉积,铁聚集区可见神经元丢失以及坏死[9],大脑皮层、基底节、齿状核及小脑均出现神经变性样变。尸检研究发现胰腺内分泌部大量铁沉积,伴胰岛β细胞显著减少[10],这也是胰岛素依赖型糖尿病出现的原因。肝活检显示肝脏结构基本正常,无肝硬化或纤维化,但肝细胞和网状内皮细胞可见铁沉积。铁离子是超顺磁性物质,且细胞内的铁造成局部磁场的不均匀,从而加快质子失相位,导致横向弛豫时间(T2)缩短,因此铁离子沉积会引起受累组织在T2WI或T2*WI上信号减低,且减低程度与铁沉积量呈正相关。与既往研究相符,影像学结果表明本例患者双侧基底节区、肝脏及胰腺存在重度铁质沉积。
CP基因位于3q24-q25,共19个外显子,现已发现40多个致病性突变,无热点突变,基因型与临床表型缺乏显著相关性[11]。本例患者携带p.R215X无义突变,铜离子结合相关6个空间结构域均受破坏,推测该突变可通过影响铜离子结合进而导致疾病的发生[12-13]。后续可针对该突变进行铁氧化酶活性检测,进而对该突变致病机制予以进一步更为完善的探索[14]。2020年新英格兰杂志报告一例患者于CP基因携带p.R215X和p.G195R复合杂合变异,其症状与本例患者相似,但无相关突变的功能学验证[15]。该患者中年女性,1型糖尿病史40年,认知功能减退2年,以反应迟钝、呼之不应于急诊就诊,伴慢性贫血及糖尿病性视网膜变性;MRI示T2加权像弥漫性、对称性低信号,基底节、丘脑、齿状核及大脑半球皮质表面存异常磁敏感信号;血清铜蓝蛋白水平低于检测下限,铁蛋白水平升高,总铁结合力降低。而我们的患者在诊断1型糖尿病26年后出现中枢神经系统症状,但并无明显贫血或视网膜变性,可见基因型与表型之间存在较大的异质性。
铁的体内调节或代谢障碍普遍存在于神经变性性疾病中,无论是NIBP,还是散发的阿尔茨海默病、帕金森病、多发性硬化,或继发性的表面铁沉积症,人们都试图通过铁螯合剂减少组织内蓄积的铁[16]。铁螯合剂主要有去铁胺(deferoxamine)、去铁酮(deferiprone)和地拉罗司(deferasirox)。在血浆铜蓝蛋白缺乏症中,目前还缺乏大样本的随机对照研究,对于螯合剂的去铁功能也尚无确凿的临床依据。去铁酮和地拉罗司降低铁蛋白水平以及肝脏和心脏铁负荷,改善贫血和糖尿病症状,而中枢神经系统症状难以逆转[17-19]。而作为铁螯合剂家族中唯一一个能通过血脑屏障的去铁酮,却给了人们新的希望[20],它不仅能降低铁蛋白水平,而且在疾病早期服用可能阻止中枢神经系统症状的发生[21]。
血浆铜蓝蛋白缺乏症可出现多系统铁沉积,而糖尿病的出现可能早于神经系统症状平均12.5年,在这个窗口期内,我们可以通过血浆铜蓝蛋白和铁蛋白的测定协助诊断,尤其是对于胰岛素依赖的糖尿病且胰岛细胞抗体(islet cell autoantibody)阴性,同时伴有贫血的患者[4]。如果能在神经系统症状前期诊断该病(pre-neuro-symptomatic phase),给予患者铁螯合剂治疗,不仅可减少体内铁负荷,改善糖尿病和贫血状态,也可能延缓中枢神经系统症状的发生,提高患者生存质量。
3 点评
血浆铜蓝蛋白缺乏症是一种罕见的成年发病的常染色体隐性遗传疾病,由编码铜蓝蛋白的CP基因突变引起。常见的临床特征包括糖尿病、视网膜病变、肝病和铁沉积所致的中枢神经系统症状。神经影像学表现为受累区域神经元丢失、铁离子沉积,其中以基底节受累最严重。实验室检查主要为血清中铜蓝蛋白极低或检测不到、铁蛋白水平升高3~40倍,血清铜、尿铜含量正常,肝铜浓度亦正常。当成年人出现原因不明的糖尿病和贫血时建议进行血浆铜蓝蛋白、铁蛋白和转铁蛋白饱和度的检测,对于早期诊断血浆铜蓝蛋白缺乏症非常有效。铁螯合剂的治疗效果,还有待于大样本的随机对照研究进一步明确。
