Tunable construction of transition metal-coordinated helicene cages
2021-03-14YongleDingChengshuoShenFuweiGnJinghoWngGuoliZhngLinglingLiMouhiShuBngshngZhuJenneCrssousHuiinQiu
Yongle Ding,Chengshuo Shen,Fuwei Gn,Jingho Wng,Guoli Zhng,Lingling Li,Mouhi Shu,Bngshng Zhu,∗,Jenne Crssous,Huiin Qiu
a School of Chemistry and Chemical Engineering,Frontiers Science Center for Transformative Molecules,State Key Laboratory of Metal Matrix Composites,Shanghai Jiao Tong University,Shanghai 200240,China
b Instrumental Analysis Center,Shanghai Jiao Tong University,Shanghai 200240,China
c Univ Rennes,Institut des Sciences Chimiques de Rennes,UMR CNRS 6226,Campus de Beaulieu,Rennes 35042,France
Keywords:Helicene Coordination cage Chiral Global helicity Circularly polarized luminescence
ABSTRACT We report a facile and tailored method to prepare globally twisted chiral molecular cages through tunable coordination of bis-bipyridine-terminated helicene ligands to a series of transition metals including Fe(II),Co(II),Ni(II) and Zn(II).This system shows an efficient remote transfer of stereogenecity from the helicene core to the bipyridine-metal coordination sites and subsequently the entire cages.While the Fe(II),Co(II) and Ni(II)-derived M2L3 (M for metal and L for ligand) cages exhibit quasi-reversible redox features,the Zn(II) analogues reveal prominent yellow circularly polarized luminescence.Interestingly,with the addition of Na2SO4,the Zn2L3 cages reassemble into sextuple-stranded Zn6L6(SO4)4 cages in which three Zn2L2 units are bound together by four sulfates and further coalesced by offset inter-ligand π-π interactions.
Molecular cages with well-defined structures and internal cavities have raised abundant interest in recent years [1,2].The synthesis of these intrinsic hollow species normally involved tailored connections of molecular building blocks either through covalent bonding or coordination to metals [3–9].The flexible linkage chemistry along with the diversity in building blocks has favored an unprecedentedly wide access to cages with tunable geometries and functions [10–12].Amongst,chiral molecular cages have drawn significant attentions due to their potential applications in chiral sensing,chiral separation and asymmetric catalysis [13–17].So far,a series of chiral molecular building blocks have been adopted to enantioselectively create these chiral entities [18,19].However,the facile construction of chiral cages with highly extended,globally distorted unsymmetrical structures remains a great challenge,majorly as a consequence that the conventional chiral molecular building blocks normally fail to efficiently transfer the local chirality to the whole cage skeletons in spite of a few advances (mainly in helicates) achieved by axially chiral building blocks [20–22].
Helicenes are a type of chiral molecules constituted byorthofused aromatic rings with unique helical shapes [23–25].Helicenecored molecule generally exhibits an apparent three-dimensionally twisted structure with chirality effectively expressed across the whole molecule along with the remote substituents [26,27].Normally,carbo[n]helicenes with the number of rings (n) larger than 5 possess stable configuration against racemization [28].We have previously demonstrated that helicene derivatives can serve as building blocks to construct covalent organic cages,which presented a globally distorted,triple-stranded helical structure with high recognition ability towards chiral guest molecules [29].Cleveret al.further synthesized bis-pyridine-terminated helicenes and used them as ligands to prepare a series of Pd-coordinated chiral cages,which also showed promising guest recognition features[30].However,the structure and function of these helicene cages is so far limited by the diversity of helicene precursor and linkage chemistry.Herein,we introduce two 2,2′-bipyridine moieties onto the 4,13-positions of [6]helicene through two alkyl bridges and develop a globally bent ligand,which favors a tunable coordination to a variety of transition metals with efficient transfer of stereogenecity to the coordination sites,and hence allow the formation of coordination cages with unique triple-stranded binuclear or sextuple-stranded hexanuclear structures and distinctive properties (Scheme 1).
We first synthesized the helicene ligand through a Sonogashira coupling reaction between 5–bromo-2,2′-bipyridine and 4,13-diethynyl[6]-helicene in both racemic and enantiopure forms(Scheme S1 in Supporting information).The resulting ligand showed a bent,non-planar structure with two extended bipyridine arms organized in a→Δ(for bis-bipyridine-terminated (P)-helicene,i.e.,(P)-ligand) or(for bis-bipyridine-terminated (M)-helicene,i.e.,(M)-ligand) chiral fashion (the dihedral angle between two arms wasca.165°,Figs.S4–1 and S4–15 in Supporting information) [31].We then mixed the ligand with Fe(CF3SO3)2in a ratio of 3:2,and subsequently observed an immediate change of color from paled-yellow to deep-red,corresponding to a fast chelating process between the bipyridine groups and Fe(II) [32].The collected product showed a clean and simple1H NMR pattern with the same number of proton environments as the ligand (Fig.1a,Figs.S2–1,S3–1 and S3–2 in Supporting information).Notably,protons H-22 and H-28 adjacent to the nitrogen atoms drastically shifted upfield from 8.98 to 8.73 ppm to 7.92 and 7.30 ppm,respectively,demonstrating the complexation between the bipyridine modules and Fe(II) [33].Electrospray ionization high resolution mass spectrometry (ESI-HRMS) revealed three main peaks atm/z=541.3949(z=4),771.5036 (z=3) and 1231.7511 (z=2),which can be indexed as [Fe2L3]4+,[Fe2L3(CF3SO3)]3+and [Fe2L3(CF3SO3)2]2+,respectively (L for the helicene ligand,Fig.1b).
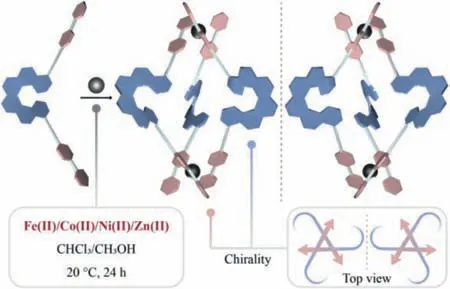
Scheme 1.Schematic representation for the synthesis of helicene-derived chiral coordination cages.
Single-crystal X-ray diffraction further confirmed the yield of Fe2L3coordination cages (Fig.1c),in which three helicene ligands were bridged by two Fe atoms (distance ofca.18.3 ˚A) and formed a triple-stranded helical structure (Fig.1d).The three surrounding helicene ligands created a sizeable internal cavity,which could accommodate a sphere with a diameter ofca.9 ˚A (with its surface just attaching the van der Waals surface of the cage framework).For the cage derived from the (P)-ligand (i.e.,(P)-Fe2L3),both two Fe-bipyridine coordination sites were inΔstereogenecity,and the whole cage framework adopted aPhelical conformation [34].Similarly,in the enantiomeric mirror series,the cage formed by the(M)-ligand (i.e.,(M)-Fe2L3) displayed anMhelical shape with two Fe-bipyridine sites solely inΛstereogenecity.It seemed that the helicene core substantially dominates the stereogenecity of the coordination domains as well as the helicity of the entire cage,which renders the cage formation reaction highly diastereoselective and prevents the formation of other diastereomers.This was actually also demonstrated by the simple1H NMR spectrum,which was indicative of the absence of other isomers.Such efficient remote transfer of stereogenic information probably can be interpreted by the minimal-energy stacking of the relatively rigid,globally bent helicene ligands.
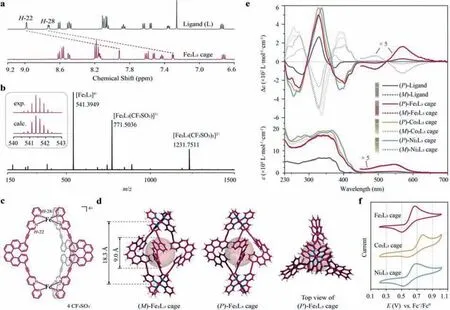
Fig.1.Fe(II)-,Co(II)-and Ni(II)-based M2L3 cages.(a) 1H NMR spectrum of (rac)-Fe2L3 cage (500 MHz,CD3CN,298 K) with comparison to (rac)-bis-bipyridine-terminated helicene ligand (L) (500 MHz,CDCl3,298 K).(b) ESI-HRMS spectrum of (rac)-Fe2L3 cage.(c) Chemical structure of Fe2L3 cage.(d) Crystal structures of (M)-and (P)-Fe2L3 cages with the largest possible spheres inside the cavity.(e) UV–vis (bottom) and ECD (top) spectra of enantiopure helicene ligands (in CH2Cl2, c=2×10-5 mol/L) and Fe2L3,Co2L3 and Ni2L3 cages (in CH3CN, c=1×10-5 mol/L).(f) Cyclic voltammograms of racemic Fe2L3,Co2L3 and Ni2L3 cages (1×10-3 mol/L in CH3CN,with 1×10-1 mol/L of n-Bu4NPF6 as the electrolyte,scan rate=50 mV/s).
The Fe2L3cage showed a group of intensive UV-vis absorption bands from 230 to 450 nm (Fig.1e),roughly reprinting the nature of the helicene ligand.The ECD signals in the region of 360–450 nm were dramatically enhanced compared to that of the helicene ligands,which was probably due to the coordination to Fe(II).The Fe2L3cage also revealed a distinctive absorption band with moderate intensity in the region of 450–650 nm,which could be attributed to the metal to ligand charge transfer (MLCT) from the Fe(II) centers to the bipyridine modules [35,36].The corresponding ECD curves were resolved in a bisignate fashion.The (P)-Fe2L3cage displayed a positive Cotton effect at 570 nm and a negative one at 490 nm,which is in accordance with theΔconfiguration of the Fe-bipyridine coordination sites [35].Meanwhile,the(M)-Fe2L3cage presented a mirror-imaged ECD curve,indicating aΛconfiguration.
We further switched the transition metals to Co(II) and Ni(II)and obtained yellow-colored products.ESI-HRMS analysis (Figs.S3–4 and S3–5 in Supporting information) and/or single-crystal X-ray diffraction (Fig.S4–4 in Supporting information) confirmed the formation of Co2L3and Ni2L3cages in a triple-stranded helical shape with the selective formation of stereogenic coordination sites analogous to the Fe2L3cage.The Co2L3and Ni2L3cages revealed similar UV-vis and ECD spectra to the Fe2L3cage in the shorter wavelength region (<450 nm),but in the longer wavelength region (>450 nm) no obvious absorption band was detected (Fig.1e),indicating a distinct metal to ligand electron transition mechanism [35].Besides,due to the introduction of electrochemically active metal coordination sites [37,38],all these three helicene cages exhibited a single redox event at +0.68 (Fe2L3cage),+0.78 (Co2L3cage) and +0.69 V (Ni2L3cage)vs.Fc+/Fc0,respectively,which appeared to be quasi-reversible (Fig.1f).
Similarly,with the addition of Zn(II) salts into the solution of the bis-bipyridine-helicene ligand,we obtained a yellow-colored compound,which showed a simple1H NMR curve (Fig.S3–6 in Supporting information) and well-resolved ESI-HRMS peaks atm/z=546.1373 (z=4,Fig.2a).Single-crystal X-ray diffraction again confirmed the formation of Zn2L3cages with homologous triple-stranded helical topology (Fig.2b).ECD spectra further reflected the chiral fashion of the resulting Zn2L3cage in solution(Fig.2c),which was similar to the Co2L3and Ni2L3cage.Like other Zn-bipyridine coordination complexes [39],the Zn2L3cages showed yellow luminescence (λmax=580 nm,Fig.2d) under the irradiation of UV light,which was considerably bathochromically shifted compared with the blue luminescence of the helicene ligand (λmax=430 nm).As a result of the global transfer of stereogenic information throughout the whole cage,these Zn2L3cages presented prominent circularly polarized luminescence (CPL) with dissymmetric factorglumof ±0.002 at 580 nm (negative for (P)-Zn2L3cage and positive for (M)-Zn2L3cage,i.e.,with the signs in agreement with the lowest ECD-active band).
Interestingly,when treating the Zn2L3cage solution with Na2SO4powder,we obtained a green-luminescent compound with a splitting of all proton environments into two sets in the1H NMR spectrum (Fig.3b).For example,the signals of protons H-22 and H-28 at 8.56 and 8.01 ppm were split,with one set drastically shifted upfield to 7.52 and 7.03 ppm and the other set moved downfield to 9.98 and 9.39 ppm,respectively.ESI-HRMS showed that this new composite possesses a relatively large molecular weight (m=4884.7608,Figs.S3–S15 in Supporting information),which can be assigned as Zn6L6(SO4)4.Single-crystal X-ray diffraction of the racemic product further illustrated a six-stranded helical structure with the integration of three tilted homochiral Zn2L2macrocyclic modules by two pairs of sulfates aligned on the vertical axis (Figs.3a and c).The intricate trigonal-bipyramid-like Zn3(SO4)2cluster also revealed a helical conformation with the two SO4vertexes slightly twisted with each other.The neighboring helicene ligands belonging to two Zn2L2modules were closely packed in a short distance ofca.3.6 ˚A and revealed unsymmetrical offsetπ-πinteractions (Figs.S4–S16 in Supporting information),which rendered a more sophisticated environment and hence the split of the1H NMR signals.This giant cage showed an expanded cavity compared with the original Zn2L3cage and could host a sphere with a diameter ofca.10.0 ˚A.
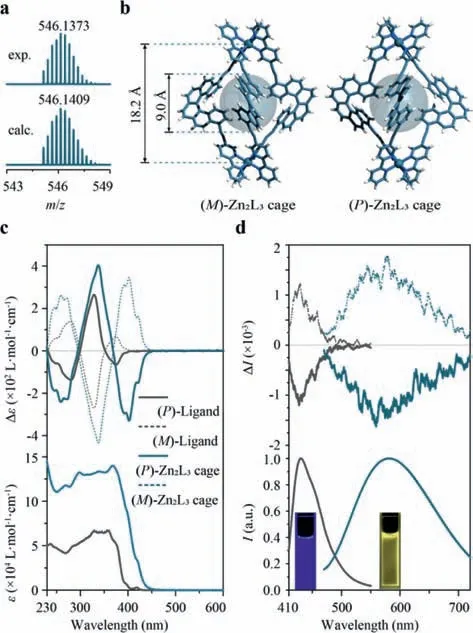
Fig.2.Zn2L3 cages.(a) ESI-HRMS and calculated mass spectra of (rac)-Zn2L3 cages.(b) Crystal structures of (M)-and (P)-Zn2L3 cages with the largest possible spheres inside the cavity.(c) UV-vis (bottom) and ECD (top) spectra of enantiopure Zn2L3 cages (in CH3OH, c=4×10-5 mol/L,compared with the helicene ligands).(d)Fluorescence (bottom) and CPL (top) spectra of enantiopure helicene ligands and Zn2L3 cages.The inset photographs show their luminescence under irradiation at 365 nm.
By gradual addition of Na2SO4into the Zn2L3cage solution in a mixture of CD3OD/CDCl3,the1H NMR spectra revealed an apparent evolution with the rising of the Zn6L6(SO4)4cage and the free ligand in a molar ratio of 1:3,and the simultaneous consumption of the Zn2L3cage precursor (Figs.S3–S16).This strongly indicated that each Zn6L6(SO4)4cage was assembled from three Zn2L3cagesvialigand exchange to sulfates,with the corresponding release of three free helicene ligands.Notably,no obvious peaks for other intermediates were observed,demonstrating that this assembly process might undergo in a cooperative pathway [40].
The Zn6L6(SO4)4cages showed similar absorption bands as the Zn2L3cages,suggesting that the major electron transition modes were generally preserved in this new coordination complex.However,the Zn6L6(SO4)4cages presented intense ECD bands in the region of 370–450 nm withΔεmax=± 1040 L mol-1cm-1,which wasca.1.6 folders of the Zn2L3cages (with consideration of the number of helicene ligands,Fig.3d).Such drastic enhancement might be resulted from the unique chiral packing of the helicene ligands as well as the twisted structure of the whole Zn6L6(SO4)4cage.On the other hand,the changes on the coordination sites apparently affected the luminescence property and hence the maximum emission hypsochromically shifted to 520 nm.Notably,the Zn6L6(SO4)4cages revealed remarkable CPL withglumof ±0.004(doubled compare to the Zn2L3cages,Fig.3evs.Fig.2d).
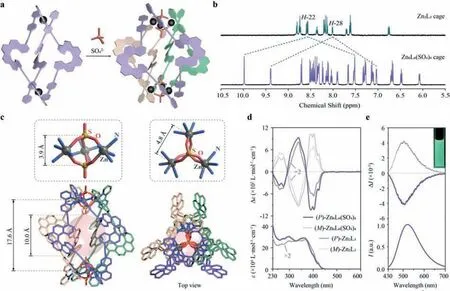
Fig.3.Sextuple-stranded Zn6L6(SO4)4 cages.(a) Schematic representation for the synthesis of Zn6L6(SO4)4 cage.(b) 1H NMR spectrum of (rac)-Zn6L6(SO4)4 cage (500 MHz,CD3OD,298 K) with comparison to (rac)-Zn2L3 cage (500 MHz,CD3OD,298 K).(c) Crystal structure of (rac)-Zn6L6(SO4)4 cage with the largest possible sphere inside the cavity,and partially enlarged views of the Zn3(SO4)2 cluster.(d) UV-vis (bottom) and ECD (top) spectra of enantiopure Zn6L6(SO4)4 (in CH3OH, c=1×10-5 mol/L) and Zn2L3 cages (the intensity for Zn2L3 cages were doubled for clearer comparison).(e) Fluorescence (bottom) and CPL (top) spectra of enantiopure Zn6L6(SO4)4 cages.The inset photograph shows the luminescence under irradiation at 365 nm.
In conclusion,we have prepared a series of chiral coordination cagesviafacile coordination of bis-dipyridine-terminated helicene ligands with a variety of transition metals.The flexible selection of metal centers substantially enriches the chemistry and functions(potentially for electrochemical or luminescence-sensitive recognition and separation of chiral guest molecules) of the resultant cages.Besides,the relatively dynamic association between the bipyridine modules and Zn(II) may also favor the fabrication of more complex chiral coordination cagesvialigand exchanges using other suitable inorganic and organic species.We are currently exploring the construction of other distinctive chiral molecular cages utilizing the diverse helicene coordination chemistry [41],aiming at innovative structures and functions.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by the National Key R&D Program of China (No.2020YFA0908100),the National Natural Science Foundation of China (Nos.92056110 and 22075180),the Science and Technology Commission of Shanghai Municipality (Nos.18JC1415500,195271040,20JC1415000).The authors thank Prof.Shunai Che’s group in Tongji University for assistance with CPL measurement.
Supplementary materials
Supplementary material associated with this article can be found,in the online version,at doi:10.1016/j.cclet.2021.05.033.
杂志排行

Chinese Chemical Letters的其它文章
- Progress in mechanochromic luminescence of gold(I) complexes
- Spinel-type bimetal sulfides derived from Prussian blue analogues as efficient polysulfides mediators for lithium-sulfur batteries
- Alopecuroidines A-C,three matrine-derived alkaloids from the seeds of Sophora alopecuroides
- Boronic acid-containing diarylpyrimidine derivatives as novel HIV-1 NNRTIs:Design,synthesis and biological evaluation
- Diaminodiacid bridge improves enzymatic and in vivo inhibitory activity of peptide CPI-1 against botulinum toxin serotype A
- Peptide stapling with the retention of double native side-chains