Boronic acid-containing diarylpyrimidine derivatives as novel HIV-1 NNRTIs:Design,synthesis and biological evaluation
2021-03-14FengFenjuWeiYnyingSunPremPrkshShrmToZhngHoLinBrijeshRthiErikDeClerqChristophePnneouqueDongweiKngPengZhnXinyongLiu
D Feng,Fenju Wei,Ynying Sun,Prem Prksh Shrm,To Zhng,Ho Lin,Brijesh Rthi,Erik De Clerq,Christophe Pnneouque,Dongwei Kng,e,*,Peng Zhn,d,*,Xinyong Liu,d,*
a Department of Medicinal Chemistry,Key Laboratory of Chemical Biology (Ministry of Education),School of Pharmaceutical Sciences,Cheeloo College of Medicine,Shandong University,Ji’nan 250012,China
b Laboratory for Translational Chemistry and Drug Discovery,Department of Chemistry,Hansraj College,University of Delhi,Delhi 110007,India
c Rega Institute for Medical Research,Laboratory of Virology and Chemotherapy,K.U.Leuven,Leuven B-3000,Belgium
d China-Belgium Collaborative Research Center for Innovative Antiviral Drugs of Shandong Province,Ji’nan 250012,China
e Suzhou Research Institute,Shandong University,Suzhou 215123,China
Keywords:HIV-1 NNRTIs NNIBP DAPY Boronic acid Molecular dynamics simulation
ABSTRACT Drug resistance remains to be a serious problem with type I human immunodeficiency virus(HIV-1)nonnucleoside reverse transcriptase inhibitors (NNRTIs).A series of novel boronic acid-containing diarylpyrimidine (DAPY) derivatives were designed via bioisosterism and scaffold-hopping strategies,taking advantage of the ability of a boronic acid group to form multiple hydrogen bonds.The target compounds were synthesized and evaluated for their anti-HIV activities and cytotoxicity in MT-4 cells.Compound 10j yielded the most potent activity and turned out to be a single-digit nanomolar inhibitor towards the HIV-1 IIIB[wild-type(WT)strain],L100I and K103N strains,with 50%effective concentration(EC50)values of 7.19–9.85 nmol/L.Moreover,10j inhibited the double-mutant strain RES056 with an EC50 value of 77.9 nmol/L,which was 3.3-more potent than that of EFV(EC50=260 nmol/L)and comparable to that of ETR(EC50=32.2 nmol/L).10j acted like classical NNRTIs with high affinity for WT HIV-1 reverse transcriptase (RT) with 50% inhibition concentration (IC50) value of 0.1837 μmol/L.Furthermore,molecular dynamics simulation indicated that 10j was proposed as a promising molecule for fighting against HIV-1 infection through inhibiting RT activity.Overall,the results demonstrated that 10j could serve as a lead molecule for further modification to address virus-drug resistance.
With the morbidity and mortality increasing rapidly worldwide,the acquired immunodeficiency syndrome (AIDS) keeps to be one of the most serious global public health problems,and the type I human immunodeficiency virus (HIV-1) is the main causative agent for it [1].In the life cycle of HIV-1,the reverse transcriptase (RT) utilizes RNA genome to synthesize doublestranded viral DNA,which was one of the most important enzymes in viral infection and a major target for antiretroviral therapies[2,3].RT inhibitors can be divided into the nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs).NNRTIs distort the enzyme’s active site and perturb the alignment of the primer terminus by interacting in a noncompetitive manner with the allosteric site(NNRTIs binding pocket,NNIBP)about 10 angstroms distant from the polymerase active site of RT,resulting in inhibition of the reverse transcription step of HIV-1 replication [4,5].NNRTIs are a component of highly active antiretroviral therapy (HAART)regimens used to fight HIV-1 due to their potent antiviral activity,high selectivity and modest toxicity [6].
Up to now,six NNRTIs have been approved by the U.S.Food and Drug Administration (FDA).Nevirapine (NVP),delavirdine (DLV)and efavirenz (EFV) constitute the first-generation,but the rapid emergence of drug-resistance and toxicities limit their clinical use[7].Etravirine(ETR),rilpivirine(RPV)and doravirine(DOR)are the second-generation NNRTIs(Fig.1),in which ETR and RPV belong to the diarylpyrimidine (DAPY) family and exhibit a broad spectrum of activity against the clinically relevant NNRTIs-resistance strains.However,their clinical application has also been complicated by emergence of drug-resistance mutations (such as E138K and RES056) [7].Besides,multiple lipophilic aromatic rings in their structure led to their poor solubility and lower oral bioavailability[7].In 2017,elsulfavirine was marketed as a prodrug in Russia for the treatment of HIV,which displayed antiviral nanomolar potency[EC50wild-type (WT) HIV-1 strain (IIIB)=1.2 nmol/L][7,8].Therefore,it is imperative to discover novel NNRTIs with improved drug resistance profiles and favorable solubility.
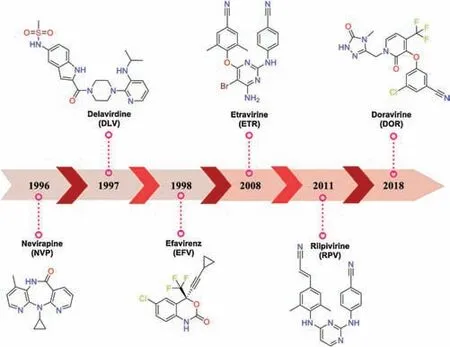
Fig.1.Chemical structures of six NNRTIs approved by the U.S.FDA.
Our previous studies have discovered a series of novel DAPY derivatives with more potent activity against the WT and mutant HIV-1 strains than the approved drug ETR [9–14].Especially,the two piperidine-substituted thiophene[3,2-d]pyrimidine derivatives,K-5a2 and 25a(Fig.2A),exhibited highly effective anti-HIV-1 activities and improved resistance profiles [15].The co-crystal structure of HIV-1 RT in complex with K-5a2 confirmed the“fourpoint pharmacophore model” of DAPY in NNIBP,which includes the hydrophobic domain,the hydrogen bonding domain,tolerant region I and tolerant region II (Fig.2B) [15].Specifically,the interactions between K-5a2 and RT were elucidated.As depicted in Fig.2C,the central thiophene[3,2-d]pyrimidine core could establish hydrogen bond with K101 and E138 directly or through a bridging water molecule;the piperidine nitrogen develops watermediated hydrogen bonds with K103 and P236;and the surfacepositioned sulfonamide group of the right wing developed a double-hydrogen bond with the carbonyl oxygen of K104 and the backbone nitrogen of V106.These extensive hydrogen-bonding networks between the inhibitor and the backbone of residues proved to be responsible for the stabilization of K-5a2 to the NNIBP.

Fig.2.(A)Chemical structures of our previously reported potent HIV-1 NNRTIs;(B)The co-crystal structure of K-5a2/RT complexes(PDB code:6c0J)and the four-point pharmacophore model;(C) The detailed interactions with WT RT of K-5a2.
Similar to the sulfonamide group,the boronic acid group proves to have the distinct ability to form hydrogen bonds [16,17].Recently,boronic acid have been extensively used for the purposes of biological probe development and drug discovery [18],especially for antiviral drugs,such as HIV-1 protease and hepatitis C virus (HCV) non-nucleoside polymerase (NS5B) inhibitors[16,19,20].In boronic acid,there are two hydroxy groups acting as both donors and acceptors of hydrogen bonds.The hydroxy groups display four lone pairs and two hydrogen-bond donors,which provide six opportunities to form hydrogen bonds with one group [16].Inspired by this,we tried boronic acid to replace the sulfonamide group in the lead K-5a2 by utilizing the bioisosterism strategy in this work.We anticipated that the hydroxy groups of boronic acid could form interactions with K104 and V106.Meanwhile,bioisosterism and scaffold-hopping strategies were also employed to displace the center core of K-5a2 with different fused pyrimidine rings and yielded twelve novel boronic acidcontaining DAPY derivatives [21].Moreover,the privileged 4-cyanovinyl-2,6-dimethylphenyl structure was introduced to the left wing of the most potent inhibitor 5j (Fig.3).The detailed structure-activity relationship(SAR)study of the derivatives and molecular dynamics simulation were also studied.
The synthetic protocols for the newly designed compounds 5a–k and 10j are outlined in Schemes 1 and 2,respectively.All derivatives were prepared by well-established methods as described in our previous articles[10–13].Commercially available 2,4-dichloropyrimidines 1a–k were selected as the starting materials,which were reacted with 4-hydroxy-3,5-dimethylbenzonitrile or 4-hydroxy-3,5-dimethylbenzaldehyde through nucleophilic substitution to afford 2a–k and 6j.Then,6j was treated with(EtO)2P(O)CH2CN in the presence of t-BuOK to give 7j.Next,2a–k or 7j were reacted with 4-amino-1-Boc-piperidine to afford 3a–k or 8j via Buchwald-Hartwig reaction.Removal of the Boc protecting group gave the corresponding key analogues 4a–k or 9j,which led to the desired compounds by nucleophilic substitution with (4-(chloromethyl)phenyl)boronic acid.
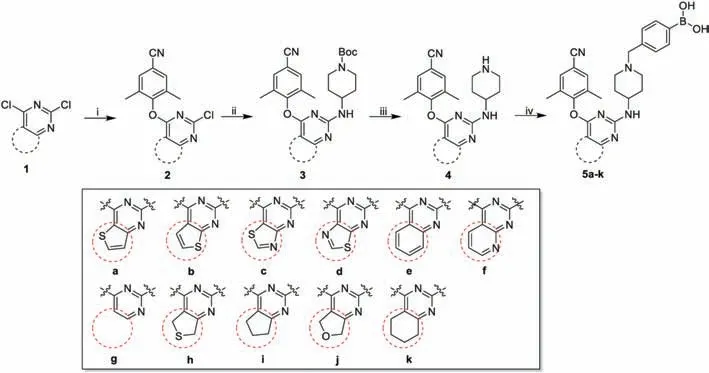
Scheme 1.Synthesis of 5a-k.Reagents and conditions:i)3,5-dimethyl-4-hydroxybenzonitrile,K2CO3,DMF,r.t.;ii)BINAP,Pd2(dba)3,Cs2CO3,1,4-dioxane,90°C,N2;iii)TFA,DCM,r.t.;iv) (4-(chloromethyl)phenyl)boronic acid,K2CO3,DMF,r.t.[(Scheme_2)TD$FIG]

Scheme 2.Synthesis of 10j.Reagents and conditions:i) 4-hydroxy-3,5-dimethylbenzaldehyde,K2CO3,DMF,r.t.;ii) (EtO)2P(O)CH2CN,t-BuOK,THF,0°C to r.t.;iii)BINAP,Pd2(dba)3,Cs2CO3,1,4-dioxane,90°C,N2;iv) TFA,DCM,r.t.;v)(4-(chloromethyl)phenyl)boronic acid,K2CO3,DMF,r.t.
The antiviral potency of the synthesized compounds was evaluated in cultured MT-4 cells infected with WT HIV-1 strain(IIIB) or a panel of NNRTIs-resistant single and double mutant strains,including L100I,K103N,Y181C,Y188L,E138K,F227L+V106A and K103N+Y181C (RES056).ETR and EFV were selected as control drugs.The values of 50%effective concentration(EC50,anti-HIV potency),50% cytotoxicity concentration (CC50,cytotoxicity) and selectivity index (SI,CC50/EC50ratio) are summarized in Table 1 and Table S1 (Supporting information).
As shown in Table 1,compounds containing sulfur atom in the central ring (5a-d)exhibited nanomolar antiviral activity against HIV-1 IIIB,L100I,K103N,Y181C,Y188L and E138K.For the K103N mutation,5a-d displayed the most potent activities with EC50values of 6.19,8.18,11.3 and 14.6 nmol/L,they were 12-,9-,7-,and 5-fold more potent than the reference drug EFV (EC50=75.4 nmol/L) but a little bit lower than ETR (EC50=3.33 nmol/L).However,all derivatives showed weaker efficacy against the double-mutant strain RES056.

Table 1 Anti-HIV activity and cytotoxicity of 5a–k and 10j.
With the aim to effectively occupy the tolerant region II and establish more interactions with NNIBP,the five-membered thiophene ring of K-5a2 was changed to six-membered aromatic and alicyclic rings,yielding derivatives 5e-g and 5h-k.Among them,5e-g turned out to be nanomolar inhibitors of HIV-1 IIIB,K103N and E138K,with EC50values ranging from 2.73 nmol/L to 24.5 nmol/L.In addition,compound 5g,with a single pyrimidine central core,displayed extremely potent activity against HIV-1 IIIB(EC50=2.73 nmol/L),which was comparable to that of ETR(EC50=3.13 nmol/L) and EFV (EC50=2.49 nmol/L).Compound 5e with the quinazoline central core showed acceptable activity against HIV-1 IIIB and K103N (EC50=6.06 and 16.2 nmol/L,respectively),and introducing nitrogen atom at the C-8 position of the quinazoline core led to a similar potency (5f,EC50=6.53 and 12.2 nmol/L,respectively).However,these compounds still showed low activities against the double-mutant strain RES056.5i exhibited high potency against WT HIV-1(EC50=4.87 nmol/L)and the single mutant K103N(EC50=5.98 nmol/L),being equipotent to that of ETR (EC50=3.13 and 3.33 nmol/L,respectively).Moreover,5k displayed moderate activity (EC50=15.1–71.8 nmol/L) against L100I,Y181C,Y188L and E138K.Introduction of sulfur or oxygen atoms at the C-6 position of 5i yielded compounds 5h and 5j,among which,5h displayed moderate potency against HIV-1 IIIB and K103N(EC50=6.60 and 10.2 nmol/L,respectively).Compound 5j was also effective against IIIB and K103N (EC50=7.01 and 7.11 nmol/L).Moreover,it turned out to be an effective inhibitor with EC50values of 13.6-47.9 nmol/L against the other mutant strains,except for RES056.
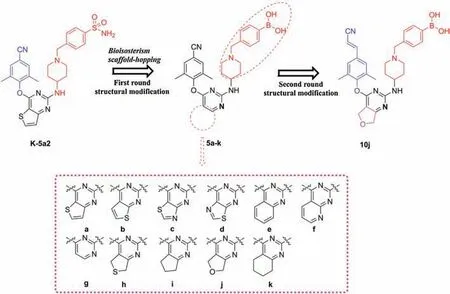
Fig.3.Design and optimization of the boronic acid-containing DAPY derivatives.
Although compounds 5j and 5i exhibited potent activities against most mutant HIV-1 strains,their activities against the double-mutant strain RES056 (EC50=1262 and 861 nmol/L) need to be improved.Our previous research has demonstrated that dihydrofuro[3,4-d]pyrimidine was a privileged scaffold for improving the anti-HIV-1 activity and physiochemical properties[13].So,the cyano group in the left wing of 5j was replaced by cyanovinyl to obtain derivative 10j,with the aim to establish more stronger interactions with highly conserved residues F227 and W229.As shown in Table 1,10j showed good inhibition against L100I with an EC50value of 9.85 nmol/L,which was about 3.8-fold more potent than 5j(EC50=37.7 nmol/L).Especially,it is noteworthy that 10j inhibited the double-mutant F227L+V106A strain and RES056 with EC50values of 43.2 and 77.9 nmol/L,which were 3.4-and 16.2-fold more potent than those of 5j (EC50=146 and 1262 nmol/L).Besides,10j showed acceptable antiviral activity against other viral strains,it was equipotent (IIIB,EC50=7.23 nmol/L;K103N,EC50=7.19 nmol/L;Y181C,EC50=19.7 nmol/L;Y188L,EC50=39.9 nmol/L;E138K,EC50=17.3 nmol/L) with 5j(EC50=7.01,7.11,20.4,47.9 and 13.6 nmol/L).These results verified that introducing the cyanovinyl group in the left wing of 10j remarkably increased the antiviral activities,especially against the double-mutant strains.Moreover,10j demonstrated higher SI values towards HIV-1 IIIB and all the tested mutant strains(Table S1).
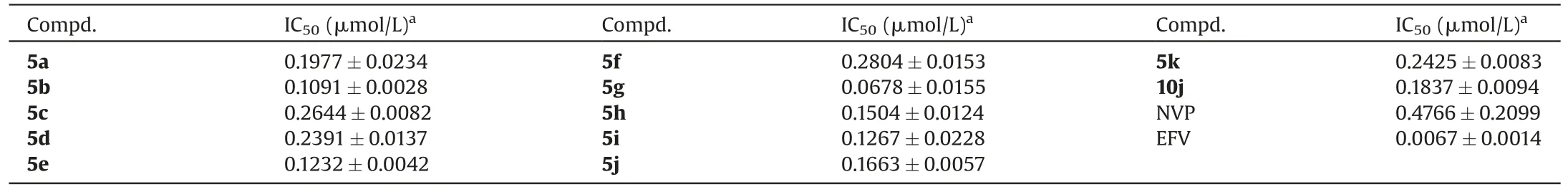
Table 2 Inhibitory activity against WT HIV-1 RT.
In order to confirm their binding target,all derivatives were evaluated for their inhibitory activity against recombinant WT HIV-1 RT enzyme(Table 2).All compounds showed high bindingaffinity with WT HIV-1 RT (IC50=0.0678–0.6585 μmol/L) as compared to that of NVP (IC50=0.4766 μmol/L).Among these,compound 5g,with a pyrimidine scaffold,exhibited the highest RT inhibitory activity(IC50=0.0678 μmol/L),which was in accordance with the anti-HIV-1 potency.These results suggest that the newly synthesized derivatives behave as typical NNRTIs.
Furthermore,we carried out the molecular dynamics analysis of 10j–RT complexes (Supporting information).The results were substantiated with molecular dynamics simulation for 100 ns that indicated the stability of all three protein structures with <0.5%outlier residues in the Ramachandran plot(Fig.S2 and Table S4 in Supporting information).The boronic acid group of the compound 10j displayed H-bond interaction to K223 residue for wildtype RT while the similar interaction was shown with K104 for mutant K103N/Y181C RT (Fig.S5 in Supporting information).Therefore,the boronic acid group helps formation of multiple hydrogen bonds to improve the antiviral activity of the compounds.
In this study,a series of boronic acid-containing diarylpyrimidine derivatives were designed,synthesized and evaluated for their anti-HIV-1 potency.The results demonstrated that compounds 5j and 5i displayed nanomolar activities against HIV-1 IIIB and single-mutant strains (EC50=4.87-71.8 nmol/L),while their activities against the double-mutant strains F227L+V106A and RES056 decreased sharply.Equipment with a cyanovinyl group at the left wing of 5j yielded the most potent inhibitor 10j with EC50values of 7.23 nmol/L (IIIB),9.85 nmol/L (L100I),7.19 nmol/L(K103N),19.7 nmol/L (Y181C),39.9 nmol/L (Y188L),17.3 nmol/L(E138K),43.2 nmol/L (F227L+V106A) and 77.9 nmol/L (RES056),which was comparable with those of ETR.The HIV-1 RT enzyme activity inhibition assay suggest that 10j (IC50=0.1837 μmol/L)acted as a typical NNRTIs.Finally,based on molecular dynamics simulation,we propose the molecule 10j promising for fighting against HIV-1 infection through inhibiting RT activity.Consequently,the compound 10j,the best inhibitor of this series,could serve as a lead for further modification to get more potent NNRTIs of important practical significance.
Declaration of competing interest
The authors report no declarations of interest.
Acknowledgments
We gratefully acknowledge financial support from the National Natural Science Foundation of China (Nos.81973181,81903453),Shandong Provincial Natural Science Foundation (No.ZR2019BH011),Natural Science Foundation of Jiangsu Province(No.BK2019041035),China Postdoctoral Science Foundation(Nos.2019T120596,2018M640641),Science Foundation for Outstanding Young Scholars of Shandong Province (No.ZR2020JQ31),Science Foundation for Excellent Young Scholars of Shandong Province(No.ZR2020YQ61),National Science and Technology Major Projects for "Major New Drugs Innovation and Development"(2019ZX09301126),Shandong Provincial Key Research and Development Project (Nos.2017CXGC1401,2019JZZY021011),Foreign cultural and educational experts Project (No.GXL20200015001),the Taishan Scholar Program at Shandong Province and KU Leuven(No.GOA 10/014).
Appendix A.Supplementary data
Supplementarymaterialrelatedtothisarticlecanbefound,inthe online version,at doi:https://doi.org/10.1016/j.cclet.2021.02.033.
杂志排行

Chinese Chemical Letters的其它文章
- Progress in mechanochromic luminescence of gold(I) complexes
- Spinel-type bimetal sulfides derived from Prussian blue analogues as efficient polysulfides mediators for lithium-sulfur batteries
- Alopecuroidines A-C,three matrine-derived alkaloids from the seeds of Sophora alopecuroides
- Diaminodiacid bridge improves enzymatic and in vivo inhibitory activity of peptide CPI-1 against botulinum toxin serotype A
- Peptide stapling with the retention of double native side-chains
- Synthesis of DNP-modified GM3-based anticancer vaccine and evaluation of its immunological activities for cancer immunotherapy