蛋白–配体复合物的结构及其相互作用分子机制的虚拟仿真实验项目的建设与教学实践
——COVID-19肺炎疫情防控时期线上教学的案例
2021-03-08万坚任彦亮饶立魏林李永健邓阳孟祥高原弘
万坚,任彦亮,饶立,魏林,李永健,邓阳,孟祥高,原弘
华中师范大学化学学院,化学国家级实验教学示范中心,武汉 430079
结构化学是在原子、分子的水平上研究微观物质运动规律,微观物质结构以及其与化学性质之间关系的科学,是化学学科的理论基础。为了让学生更好地学习和掌握结构化学相关知识,我们构建了模块化、功能化的“结构化学课程群”[1],包括结构化学、分子模拟基础、理论与计算化学课程,进行分类分层教学,以满足学生个性化发展需求[2]。由于该课程群所涉知识大多抽象、理论性强、数理要求高,学生在学习过程中会遇到相当大的挑战。为提高学生的学习积极性,帮助学生掌握化学学科的理论基础、学科思维、学科表达和学科应用,全面培养学生终身发展所需的学习力、思考力和发展力,在长期的教学实践中,我们逐步构建、完善并实施了基于现代教学理念和理论的“四轮驱动”教学策略和方法,如图1所示。

图1 本虚拟仿真实验项目在结构化学课程群教学中的定位与作用示意图
该教学策略方法包括由点到面的知识框架图(Schema)的系统凝练;充分体现“数-形结合”能力与素养,且由浅入深的挑战性问题(Challenge Question, CQ)的探索解决;问题导向、基于课程项目、注重过程评价的“深度”教学实践(Problem Oriented,Project-Based, Process-Evaluated Teaching and Learning,P3OBE)的分组实施;以及通过学习者充分准备和参与的口头报告与研讨(Oral presentation and discussion)环节,完成上述几类“深度”学习任务,自我完善后以“微课”总结提交最终学习“成果”的综合训练。“蛋白–配体复合物的结构及其相互作用分子机制”的虚拟仿真(Virtual Simulation,VS)实验项目是“结构化学课程群”理论教学的实践和应用,既是P3OBE由浅入深、不可或缺的重要组成部分,亦是后续P3OBE进入高阶训练的线上仿真实验教学的入门环节(图2),充分体现了“能实不虚,虚实结合,虚为实用”的虚拟仿真实验三原则。虚似仿真实验网址:http://www.ilabx.com/details/v5?id=4299&isView=true。
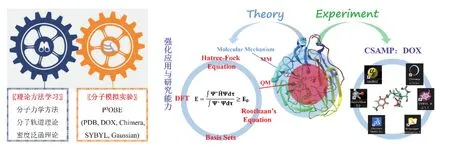
图2 “双轮驱动”的蛋白–配体结合构象精确预测的P3OBE实践学习框架图
1 实验内容
蛋白–配体结合构象是生物体系分子识别、生理生化性质与功能的分子机理、基于结构的药物设计等研究领域中的一个最基本,同时也是最重要、最复杂的关键科技问题。对蛋白–配体结合构象的研究与结构化学课程群强调的“宇内万物由物质组成,相互作用决定其结构,结构决定其性质与功能”的学科思想高度契合。蛋白环境及其相互作用决定了蛋白–配体的结合构象及其结合能力强弱,而蛋白–配体结合构象和结合能力又决定了该配体的生物活性的大小。目前获得蛋白–配体结合构象最可靠的实验方法是X射线晶体衍射结构分析,为此本实验内容包括了:X射线衍射法测定晶体结构的基本原理以及晶体学的相关基础知识;蛋白质和配体小分子相互作用的主要类型和特征,蛋白–配体复合物的主要功能——催化与抑制催化的机理。受制于蛋白质的复杂性(分子量大、纯化表达困难、晶体培养条件苛刻、晶体分辨率低等),通过实验手段解析蛋白质及其配体复合物结构信息往往成本高、难度大。运用现代分子模拟方法对蛋白–配体的结合构象进行理论计算预测无疑是对上述实验方法的一个有益、重要且必须的补充,同时也是结构化学研究中的重难点内容。因此,学习和掌握蛋白–配体结合构象理论计算的基本原理和步骤是本虚拟仿真实验的第三个研究内容。目前应用最广泛的蛋白–配体结合构象计算方法是分子对接,该方法速度快但准确性不高,这是由于分子对接在计算时为了节省计算量而忽略了电子效应。我们通过引入量子力学计算发展了DOX方法[3,4],显著地提高了蛋白–配体结合构象计算的准确性。所以,本虚拟仿真实验教学亦是我们相关科研成果的转化。在本实验中,学生将同时体验和比较X射线晶体衍射结构分析、分子对接和DOX,深入理解“微观物质结构由微观物质运动法则即量子力学决定,而微观物质结构又决定其在宏观世界的功能”这一结构化学基本理念,并强化对学科知识和学科应用的掌握。同时,通过上述虚拟仿真实验项目的探究学习也为“结构化学课程群”后续系统的 P3OBE实践打下较为坚实的知识与方法应用的前期基础。
2 实验原理
2.1 X射线衍射晶体结构分析
1912年,Laue将一束X射线穿过硫酸铜晶体,成功地观察到了X射线衍射图像。该实验解决了两个争论已久的问题:首先证明X射线是波长很短的波,其次证明晶体是原子有规则排列而成的固体。该实验揭开了通过X射线衍射测量晶体结构的序幕,基于此贡献,Laue在1914年获得了诺贝尔物理学奖。随后Bragg父子在Laue的基础上给出了晶体结构与衍射方向之间的定量关系,奠定了X射线晶体结构分析的基础,这就是著名的Bragg方程:2dsinθ=nλ,式中d是晶体中的晶面间距,θ是入射X射线与晶面的夹角,λ是X射线的波长。上述公式表明,当n为正整数时,若等号成立,即可观察到叠加而成的衍射光。因此,当观察到衍射光时,可根据该夹角和入射光的波长计算出晶面间距,获得晶胞的大小和形状。此外,衍射强度决定了各原子在晶胞中的位置,通过测量X射线穿过晶体时产生衍射光的衍射方向和强度,可以推算出晶体的微观结构,这就是X射线衍射晶体结构分析。
2.2 蛋白质晶体结构分析
1958年,英国科学家John Kendrew和Max Perutz最先发表了用X射线衍射得到的高分辨率肌红蛋白Myoglobin的三维结构[5],开创了蛋白质晶体结构分析技术。两位科学家也因此获得了1962年的诺贝尔化学奖。当前蛋白质晶体结构分析的基本流程是,首先通过基因克隆技术获得大量用于结晶的高纯度目的蛋白质,然后将蛋白质与预先配好的结晶溶液进行混合,利用悬滴法获得蛋白质晶体。悬滴法结晶的基本原理是通过蒸发降低蛋白质溶液中水含量,使得蛋白质结晶析出。结晶溶液中主要包括缓冲溶液、沉淀剂、离子盐。其中,缓冲溶液的作用是保持整个体系的pH稳定;沉淀剂的作用是使得体系的水含量降低,促使蛋白质结晶析出;离子盐主要用于中和蛋白表面的电荷,增加蛋白之间碰撞堆积的概率。蛋白质通过悬滴法生长2–30天达到一定尺寸后,便可捞出放入液氮中保存,送往X射线光源进行衍射实验。值得一提的是,蛋白质虽然是大分子,但晶体尺寸往往十分小。因此蛋白质晶体中结构基元总数较少,衍射光较弱。为获得高精度的结构数据,则需要使用强度极高的 X射线。同步辐射光源通过令高速运动的电子在磁场中作曲线运动来产生高强度 X射线,是进行蛋白质晶体衍射实验的理想光源。目前我国已建成的同步辐射光源中最先进的是上海同步辐射光源。
2.3 蛋白–配体相互作用
配体是指各类能和蛋白质结合的小分子,包括金属离子、辅因子、底物、抑制剂或激动剂分子等。它与蛋白质的结合作用主要来自于蛋白质原子与配体原子之间形成的氢键、离子键、静电作用和疏水作用,有时也包括共价键。蛋白–配体相互作用对蛋白质的功能起着至关重要的作用,例如酶蛋白催化反应的本质是酶蛋白通过与底物分子的相互作用促使底物转化为产物。若用另一小分子代替底物与酶蛋白结合,便可阻断蛋白-底物相互作用,起到抑制酶蛋白活性的作用,这就是抑制剂的原理。而抑制剂是当前创新药物研发的热点。以本实验中的人体 HMG-CoA还原酶(3-hydroxy-3-methyl glutaryl coenzyme A reductase,以下简称HMGR)[6]为例,HMGR在人体中负责催化从3-羟基-3-甲基戊二酰辅酶A(HMG-CoA)到甲羟戊酸(MVA)的合成反应,这一反应恰好是肝细胞合成胆固醇过程中的关键步骤。因此,抑制HMGR酶蛋白的活性可降低体内胆固醇,从而治疗高血脂及相关疾病,著名的他汀类降血脂药物,如明星药美伐他汀(Mevastatin)和洛伐他汀(Lovastatin)等,均属于HMGR抑制剂。
2.4 蛋白–配体结合构象的计算
准确解析生物大分子-配体相互作用的三维空间结构(结合构象)是生物体系分子识别、生理生化性质与功能的分子机理、基于结构的药物设计等研究领域中的一个最基本同时也是最重要、最复杂的关键科技问题。目前该领域常用的实验方法主要有晶体 X射线衍射法、高分辨液相核磁共振法(NMR),以及近几年逐渐受到关注的冷冻电镜技术。然而受制于生物大分子的复杂性(分子量大、蛋白纯化表达困难、晶体培养条件苛刻、晶体分辨率低等),通过实验手段快速解析蛋白质等生物大分子及其配体复合物结构信息的方式仍然受到限制和挑战。运用现代分子模拟方法对生物大分子-配体的结合构象进行理论预测无疑是对上述实验方法的一个有益、重要且必须的补充,同时也是理论与计算化学复杂体系与方法研究中的重要内容。理论计算研究生物大分子中配体分子基态和激发态的性质与功能的前提就是要获得准确的生物大分子与配体分子的结合构象。因此,在没有蛋白复合物晶体或高分辨 NMR等实验结构数据的情况下,发展复杂生物体系的理论计算策略与方法以获得准确的生物大分子与配体的结合构象成为该研究领域首先需要解决的关键科学问题。
2.4.1 传统蛋白–配体结合构象计算方法:分子对接
目前应用最广泛的蛋白–配体结合构象计算工具是分子对接。分子对接是通过研究配体(如药物小分子)和受体(如蛋白质或其他生物大分子)间相互作用,并预测其结合模式和亲合力的一种理论模拟方法。它是依据配体与受体作用的“锁-钥原理”(lock and key principle)模拟小分子配体与受体生物大分子间的相互作用的,这种相互作用主要包括静电作用、氢键作用、疏水作用、范德华作用等。
分子对接首先产生一个填充受体分子表面口袋或凹槽的球集,然后生成一系列假定的结合位点。依据受体表面的这些结合点与配体分子的距离匹配原则,将配体分子投映到受体分子表面,并通过旋转和扭动配体分子得到一组可能的受体-配体结合构象。之后,计算这些结合构象的亲和力,并对计算结果进行打分,按得分高低判定最优受体配体结合构象并基于该构象给出对配体与受体的结合能力的计算。分子对接的种类主要包括:(1) 刚性对接:在对接过程中,研究体系(受体和配体)的构象不发生变化。适合考察比较大的体系,如蛋白质和蛋白质之间以及蛋白质和核酸之间的对接。(2) 半柔性对接:指在对接过程中,研究体系尤其是配体的构象允许在一定的范围内变化。适合处理大分子和小分子间的对接,且在对接过程中,小分子的构象一般是可以变化的,但大分子是刚性的。(3) 柔性对接:指在对接过程中,研究体系的构象基本上可以自由变化。一般用于需要精确考虑分子间识别的情况。但是由于计算过程中体系的构象可以变化,所以柔性对接计算耗费最大。目前应用最广泛的是半柔性分子对接计算。
2.4.2 高精度蛋白–配体结合构象预测策略与计算方法:CSAMP-Strategy Based DOX Method
目前传统分子对接计算方法获广泛应用,它的优点是计算速度快,但同时也有计算精度不高的问题。根据文献中对七种主流分子对接程序的系统评测,可以发现,以预测/正确构象差异RMSD <0.2 nm为标准,不同分子对接方法的准确率大致在30%–60%,尽管RMSD < 0.2 nm的标准并不算严格。
文献报道还发现分子对接搜索构象的效率较高,在所产生的全部结合构象中往往已包含“正确”结合构象。其预测结果不准的根源在于构象排序所用的经验打分函数的精度有限。若用更高精度的理论取代经验打分函数对构象进行排序,即可提高结合模式预测的准确度。我们课题组与复旦大学徐昕教授合作,基于上述思路发展了高精度蛋白–配体结合模式计算方法DOX。DOX方法的主要特点是利用CSAMP (Conformation Search Across Multiple-Level Potential-Energy Surfaces)策略[3],将分子对接、半经验量子化学理论和第一性原理密度泛函理论进行有机结合。所谓CSAMP策略即是用低精度理论进行全局构象搜索,用中精度理论进行构象筛选,用高精度理论进行构象排序。该策略结合了低精度理论的高效率和高精度理论的准确性,从而允许我们利用较少的计算量逼近计算量无法估计的高精度搜索全势能面的结果,实现第一性原理级别的高精度构象预测。在 DOX方法中,PM7半经验方法被用于对 Surflex Dock产生的 300个构象进行筛选,取前 10个用 eXtended ONIOM(XO)计算进行最终排序。XO是徐昕教授课题组开发的复合计算化学理论方法,通过分块计算显著降低高标度理论的计算量,从而实现巨大体系的密度泛函理论计算。
在Astex标准测试集上的测试结果表明DOX方法的结合模式计算准确率(以RMSD < 0.2 nm为标准)高达99%,显著优于分子对接的表现。采用更严苛的标准(RMSD < 0.1nm),DOX方法的准确率仍可达到70%以上,这意味着DOX计算的精度已接近晶体结构。需要说明的是,DOX1.0方法已于2016年发表[4],DOX2.0方法于2019年发表[3]。目前可通过访问DOX网页服务器进行计算服务(http://202.114.32.71:10280/wandox/ DOXserver/home.html)。
3 实验方法与步骤要求
3.1 实验方法描述
原子、分子等微观结构的相互作用是化学实验的核心,但宏观世界和微观世界的尺度鸿沟令我们的肉眼无法直接观察到这些微观结构,这也为学生学习和理解相关知识设下了障碍。本实验将利用虚拟现实技术,以肺炎链球菌3-羟基-3甲基戊二酰辅酶A(HMGR)和当前世界上使用最广泛的明星降血脂药-美伐他汀为例,全面、真实、细致地让学生了解微观结构的获取方法,掌握分析研究酶蛋白与抑制剂相互作用的不同手段,从而使学生能以直观的方式观察微观结构,学习微观世界。整个实验分为三个模块:模块一为 X射线衍射晶体结构分析基础;模块二为蛋白–底物相互作用分析和操作模块;模块三为蛋白-抑制剂相互作用分析模块。在本实验教学中,学生首先学习如何将美伐他汀对接到HMGR底物活性空腔中,再运用3D互动功能寻找在分子对接计算级别下打分最高的美伐他汀结合构象与HMGR形成的氢键数量。但是与晶体的美伐他汀与HMGR形成的氢键相比,分子对接中打分最高构象与HMGR的相互作用中少了一根氢键,分子对接结果不够准确。为此,学生需要进一步学习运用最新改进的高精度DOX方法,对抑制剂在酶蛋白中的结合构象进行深度优化,最终通过UNIT的3D互动功能,学生可以发现影响抑制剂的抑制活性的氢键网络和关键药效团明显增加,并与晶体结构相比非常吻合。课后教师根据软件操作的情况,结合学生撰写的“实验分析报告”进行综合评分。
3.2 学生操作步骤
3.2.1 X射线衍射晶体结构分析基础(模块一)
衍射基础知识学习:学生可点击“X射线衍射基础知识”,观看解说动画,学习X射线衍射法测定晶体结构的基本原理。晶体样品放置:学习完射线衍射法测定晶体结构的基本原理后,学生可点击“X射线单晶衍射虚拟实验(示范)”,以CsCl晶体的X射线衍射实验为例,学习如何在显微镜中观察晶体。之后将CsCl晶体放置于X射线衍射仪上,学习调整晶体位置(图3A)、旋转样品台(图3B)、用CCD拍摄衍射光(图3C, D)等基本操作。

图3 X射线衍射仪上晶体的放置、调整、旋转及衍射光拍摄等仿真实验图
衍射结果处理:通过X射线衍射可得到晶胞的形状、各原子的相对位置及相对衍射强度。之后根据上述信息进行晶体结构解析,包括通过晶胞的形状确定晶系和点阵形式,根据原子相对位置确定其分数坐标,根据衍射强度归属元素符号。在示范实验中(图4A),每一步均有详细介绍,操作错误会收到提示(图4B)。生成实验报告:点击模块一的“实验报告在线填写”,将会生成该模块的实验目的、实验原理以及学生在实验操作过程中的各项操作的错误数和打分情况,具体如图4(C,D)所示。对于实验原理和实验目的部分,我们将在课堂上会组织学生进行口头报告和分组讨论。

图4 晶胞中各原子的相对位置和相对衍射强度的虚拟仿真操作及模块一的实验报告情况分析
3.2.2 蛋白–底物相互作用分析(模块二)
蛋白晶体衍射知识学习。学生可以点击“蛋白质X射线衍射晶体结构分析”观看动画,学习蛋白质晶体结构分析方法。配催化反应知识学习:HMGR/HMG-CoA复合物的结构与机理。蛋白质在生命过程中的作用无可替代,这也决定了对其性质、机理和功能的研究需要建立在其三维结构的基础上。前述X射线衍射晶体结构分析法正是获取蛋白质精细三维结构的主要途径。根据X射线获得的蛋白–配体复合物精细三维结构,学生将会发现和学习底物(HMGR-CoA)和辅因子在蛋白质(HMGR)活性空腔中进行催化反应的机制和原理。在学习中,学生开始认识到“酶催化反应的效率是由酶蛋白活性空腔中的关键氨基酸残基(药效团)来决定”,发现了蛋白–底物的相互作用主要是配体与药效团形成氢键作用,并掌握了决定酶HMGR催化反应效率的关键药效团。
3.2.3 蛋白–抑制剂相互作用分析(模块三)
分子对接原理的学习:在学习了影响底物催化反应机理关键药效团后,学生将在该模块中使用目前最流行的药物设计方法——分子对接进行HMGR抑制剂的设计。首先在“分子对接与DOX计算”子模块中,学生可以理论学习分子对接和DOX计算的基本原理。分子对接设计HMGR抑制剂:点击“分子对接获取 HMGR/美伐他汀的结合模式”,学生开始尝试将降血脂药美伐他汀对接到HMGR酶蛋白中,并选择HMGR的三维结构。在分子对接前需要对酶蛋白结构进行合理化的优化处理,点击“蛋白删除水分子”按钮,删除蛋白中不参与催化反应的水分子,以避免影响分子对接结果。X射线衍射无法正确分析酶蛋白中氢原子的位置,而没有氢原子的蛋白是无法进行正确的分子对接计算的,因此在对接前必须要对没有氢的酶蛋白进行加氢处理,点击“蛋白和小分子加氢”对酶蛋白进行合理的加氢操作和处理。抑制剂在蛋白中的结合模式分析:点击“提交”按钮,对抑制剂和酶蛋白的相互作用结合构象合理性进行计算分析,学生可寻找美伐他汀在 HMGR酶蛋白中的结合位置,并结合前面底物的结合模式分析美伐他汀是否竞争性地占据HMGR底物空腔。此外,还需要从整体视角观察HMGR酶蛋白活性空腔的形状,并分析抑制剂小分子与HMGR活性空腔是否形状匹配。在三维真实模式中,学生首先在分子对接的打分最高构象中,寻找美伐他汀与HMGR中的关键药效团能形成的氢键,并手动选择,如图5(A, B)所示。当然,学生也可以点击晶体中美伐他汀与HMGR中的关键药效团形成的氢键情况。通过两种结构模式的比较,学生可以发现,分子对接确实可以还原部分晶体结合模式,虽然有一个氢键在分子对接打分最高的构象中无法找到,但其实存在这样的结合模式,只是无法正确评价并打分。
DOX计算分析HMGR与抑制剂的结合模式:点击“DOX计算获取HMGR/美伐他汀的结合模式”,学生可以采用高精度计算方法(DOX)优化上述结合模式,如图5(C, D)所示。之后,观察计算所得的新结合模式,识别主要的蛋白–抑制剂相互作用。实验报告:在每个模块完成后,点击“实验报告在线填写”,将会生成对应模块的实验目的、实验原理以及学生在实验操作过程中的各项操作的错误数和打分情况。对于实验原理和实验目的部分,我们将会在课堂上组织学生口头报告和分组讨论实验原理、实验过程、实验结果、实验得分以及心得体会。

图5 在HMGR空腔中抑制剂对接构象和晶体构象的差异性及DOX计算过程的虚拟仿真示意图
4 结语
本虚拟仿真实验项目(软件著作权登记号为:2019SR1073615[7])已经在结构化学、分子模拟基础以及理论与计算化学等课程中针对我校化学类专业16和17年级部分本科生尝试开展了线上虚拟仿真实验教学服务。在2020年春季学期COVID-19肺炎疫情防控期间,响应教育部“停课不停学”的号召,本项目除了为本校的学生提供线上自主学习服务之外,也积极面向全国高校学生免费开放使用服务。目前,国家虚拟仿真实验教学项目共享平台上本实验项目浏览量已超过15,000人,实验完成人数超过500人,起到了很好的线上教学示范作用。
