煤制化学品:合成气直接制低碳烯烃催化剂研究进展
2021-03-08刘义涛朱明辉杨子旭孟博涂维峰韩一帆
刘义涛,朱明辉,杨子旭,孟博,涂维峰,韩一帆,
(1 化学工程联合国家重点实验室,华东理工大学,上海200237;2 先进功能材料制造教育部工程中心,郑州大学,河南郑州450001)
低碳烯烃(乙烯、丙烯和丁烯,C2~C4烯烃)是现代化学工业的基石,被广泛应用于染料、塑料、药物、橡胶、润滑剂和化妆品的生产原料[1]。当前,低碳烯烃主要是通过石脑油的蒸汽裂解、低碳烷烃的脱氢等方式进行生产。考虑到化石能源的枯竭以及公众对环境问题的关注,研究者对使用非石油原料通过合成气生产低碳烯烃进行了广泛的研究。间接法,如甲醇制烯烃(MTO)和二甲醚制烯烃(DMTO)[2-3],已经成功工业化。近年来,通过费-托(FT)工艺将合成气直接转化为低碳烯烃获得了广泛关注,它避免了形成中间体(如甲醇、二甲醚等)的步骤,降低了热量、水的损耗,从而降低了投资成本。该方法的最大挑战是开发具有优异选择性的高效催化剂,以满足工业化的需求。
针对以上现存问题,本文将从合成气直接法催化转化制烯烃工艺路线、CO 加氢的热力学分析、合成气直接法制低碳烯烃催化剂、CO 加氢反应机理等方面进行论述。
1 合成气催化转化制烯烃工艺路线
目前,学术界主流研发的合成气直接法制烯烃主要有两种工艺路线:一是双功能催化剂反应偶联,合成气经甲醇或甲氧基合成以乙烯、丙烯、丁烯为主的低碳烯烃;二是费-托合成制低碳烯烃,采用传统Fe、Co 基催化剂,催化CO 经费-托合成直接加氢制低碳烯烃。
1.1 双功能催化剂反应偶联
在合成气直接制低碳烯烃双功能催化剂的研发中,国内课题组近年来发表了突破性的研究成果。2016年,大连化学物理研究所包信和教授团队[4]在《科学》杂志上介绍了一种新型氧化物-分子筛双功能催化剂OX-ZEO,由金属氧化物(ZnCrOx)和多孔SAPO沸石(MSAPO)组成(图1)。在400℃、2.5MPa、H2/CO=2.5 的条件下,通过将CO 活化和C—C键形成过程分开,突破传统费-托合成产物分布的限制,烯烃选择性高达80%;同年,厦门大学王野教授团队[5]在《德国应用化学》上发表的通讯文章中报道了类似的结果。他们采用一个双功能催化剂偶联了甲醇合成和甲醇制烯烃(MTO)两个过程,ZnZrOx催化剂在400℃、H2/CO=2、1.0MPa 对甲醇和二甲醚有很好的选择性,偶联MTO 的SAPO-34分子筛催化剂,同时为了抑制烯烃加氢,减少了分子筛上Brønsted酸性位的密度,低碳烯烃选择性最高达到74%。这种反应偶联的方式得到了广泛关注[6]。
1.2 费-托合成制低碳烯烃

图1 反应偶联直接法制低碳烯烃示意图[4-5]
费-托合成制烯烃(Fischer-Tropsch to olefins,FTO)是指合成气在0~3.0MPa、250~340℃的工业费-托反应条件下,采用传统的Fe、Co基催化剂催化CO 加氢生成低碳烯烃,同时伴随甲烷、长链烃类等副产物的生成。科学家们考虑这一想法已经超过50 年了,目前已有大量的直接法制烯烃的文献报道。由图2 可知,合成气经费-托合成制烯烃的专利数量与油价成较好的正相关,石油资源短缺和高油价都促进了费-托合成制烯烃的研究。尽管2013 年后石油价格明显下跌,但是对于合成气直接法费-托合成制烯烃的研究仍然热度不减。采用煤气化制合成气为原料,以合成气制乙烯为例,费-托合成直接制烯烃工艺涉及的全流程主要反应如式(1)~式(5)。

考虑水气变换

当x=0
当x=1

即总反应

根据水气变换反应程度,该工艺碳的利用率为50%~100%,适宜氢碳比为0.5~2;FTO 反应通常在中压下进行(2MPa);无需水气变换装置,流程少,一次投资成本较低。

图2 1927—2017年“费-托合成制低碳烯烃”学术论文和专利(柱状图)与油价的关系(实线)
因此,直接法费-托合成制备低碳烯烃具有如下特点:①该工艺只包含煤气化、合成气转化以及产物分离系统,可省去甲醇合成工艺中的水气变换反应装置、甲醇分离装置、MTO反应装置等,整套生产装置流程的简化可大幅降低装置的一次性投入和能耗;②反应压力2MPa 左右,远低于甲醇合成5MPa的反应压力,降低压缩能耗;而且,直接法反应在中压压力下进行,产物除尘可采用机械过滤的方法,避开水洗,减少水耗以及污水排放,节约成本,提高环境和经济效益;③合成气单程转化率的提高减少了物料循环的能耗;④直接法具有更高的原子经济性;⑤合成气直接制烯烃工艺产品也异于传统制备路线,产品适应性强。除了乙烯和丙烯之外,所得烯烃几乎皆为附加值更高的高碳直链α-烯烃,异构烯烃含量很低。由于高碳直链α-烯烃应用更为广泛,附加值更高,一旦实现低成本合成气高选择性直接制取烯烃,将带动许多相关行业的发展。
2 CO加氢的热力学分析
CO 加氢制烃类同时生成CO2和水的过程是放热反应,例如式(6)。

放热随着产物链长的增加而增多,因此移热是一个反应器操作和过程设计的关键因素。实际上,在FTS反应器中催化剂表面上往往同时发生多个反应。表1 列出了一系列可能的反应[7]。热力学产物选择性一般遵循以下顺序:烷烃>烯烃>醇类,而且产物分布随催化剂种类和产物条件变化显著。费-托合成的主要化学方程式如式(7)。
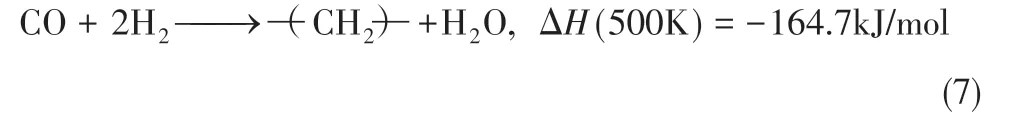
如果将反应(7)中的—(CH2)—替换为CH4,可以得到反应式(8)。
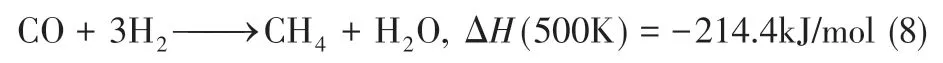
进料合成气的氢碳比取决于目标产物、操作条件和催化剂性质。当反应物处在化学平衡,Ribblett比的值趋近于1,即式(9)。
一般认为,水气变换(water gas shift,WGS)反应对CO 加氢反应计量数有一个显著的影响,如式(10)。

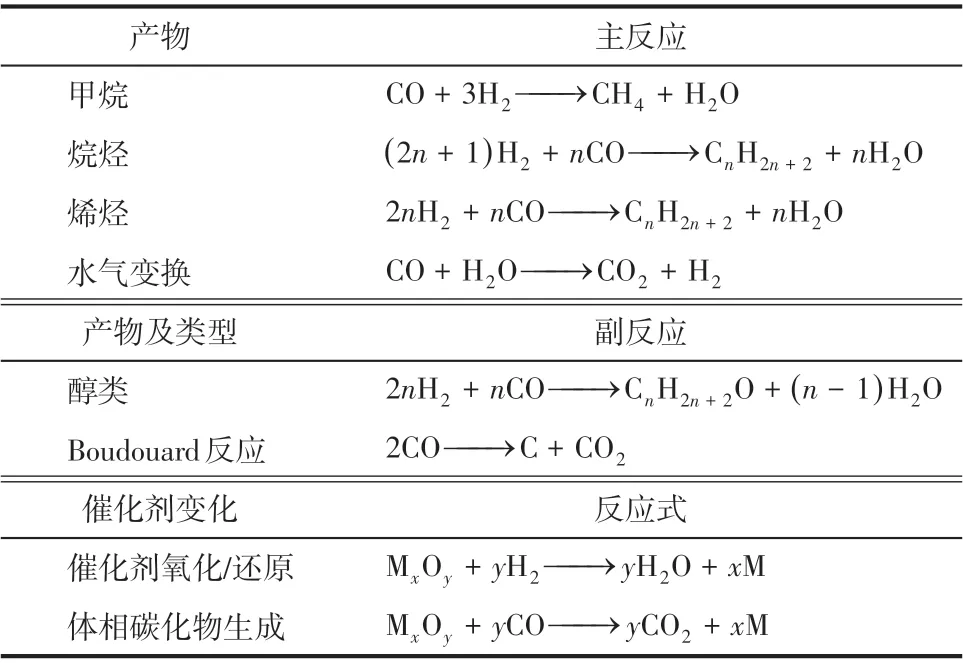
表1 CO加氢反应[7]
Co基催化剂一般需要合成气比例H2/CO≈2,实际约为2.15;Fe 基催化剂一般水气变换活性较高,实际H2/CO比约为1.7[8],取决于反应温度。对于高温费-托反应(300~350℃),WGS 能够达到平衡,并且能够促进烃类生成。例如,用H2/CO=1合成气的FTS和WGS的烃类生成净反应,如式(11)。

比较CO2和H2O 的Gibbs 自由能可以推测,在合成过程中CO2的生成是优先于水的。对于低温反应(180~250℃),WGS处于非平衡,但是CO2可能显著生成。此外,大部分和烃类生成相关的反应都伴随着碳沉积过程,如Boudouard反应,如式(12)。

对比以上主要反应,可以预测CO 加氢过程最容易生成甲烷以及大量的积炭。在200~400℃,甲烷和积炭的生成是热力学有利的,而对烃类的生成不利[9]。
3 合成气直接法制低碳烯烃催化剂
合成气直接法经费-托合成制低碳烯烃的催化剂的研发主要是围绕对传统费-托合成催化剂的改性。在CO 加氢反应中,H2和CO 在催化剂表面的吸附性能是至关重要的。如图3所示,蓝色虚线左侧的金属在金属态即具有直接解离CO 的能力,但是除Fe 元素外,其他金属在自然环境下不易形成金属态,在极为苛刻条件下才能还原出来。红色虚线右侧的金属不具有CO 解离能力,蓝色背底的金属元素一般用来做CO 加氢制甲醇或低碳醇的活性金属或助剂,提供非解离CO 插入的活性位。蓝色和红色虚线间的金属在一定条件下可以解离CO,黄色背底的金属元素被视为费-托反应制烃类产物的活性组分。众所周知,Fe、Co、Ru 基催化剂是费-托合成反应催化剂活性组分,考虑到成本问题,Fe、Co 基催化剂的实际应用更为普遍。相比于Co基催化剂,Fe更多地催化CO加氢生成烯烃和含氧化合物,可能由于Fe 元素加氢能力较弱。而且金属态Co对于CO加氢具有活性且相对稳定,但Fe 催化剂在工业费-托反应条件下容易发生相转变,形成由Fe 碳化物、Fe 氧化物组成的复杂混合物。
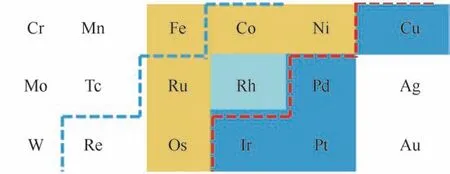
图3 过渡金属元素CO解离能力
3.1 Fe、Co基催化剂活性相
对于铁基催化剂,一般认为铁碳化合物是催化生成烃类的活性相。但由于碳化铁种类繁多,按含碳量从低到高依次为Fe3C、Fe5C2、Fe7C3、Fe2.2C、Fe2C,至今仍未对是何种碳化铁为费托合成活性相形成共识。一方面是由于碳化铁在空气中极不稳定,容易发生剧烈氧化乃至燃烧,导致难以对其结构进行精确地离线表征,而实时的原位表征又常常受制于其高温高压的反应条件,往往难以进行或者相应的信号较差;另一方面,由于各个研究往往都是在不同的条件下进行,而不同的温度和处理气氛又会控制碳势,进而影响对铁的渗碳能力,生成不同的铁碳化合物或者混合物,因此不同反应条件下所观测到的碳化铁也不尽相同。绝大部分的研究都倾向于认为Fe5C2是活性相[10]。Chang 等[11]研究了不同铁碳化合物与其催化性能之间的关系,发现在中温(260~300℃)反应条件下,Fe7C3具有最高的本征活性。Xu 等[12]报道了Fe2C 作为低温费托合成的催化剂。他们发现在150~200℃的范围内,Fe2C 是主要活性相。在没有其他任何助剂的加入下,其费托活性仍能够大大超出其他铁基催化剂。Liu 等[13]认为Fe3C 是费托合成制备低碳烯烃的活性相,在320℃的反应温度下,经锰修饰的铁基催化剂获得了60%左右的低碳烯烃。他们认为Fe3C 的弱加氢能力是促进生成烯烃的关键。由此可以看出,不同的研究之所以能够提出不同的碳化铁作为活性相,跟其具体的反应温度密切相关。
对于钴基催化剂,大部分的研究都认为单质钴为费托合成的活性相。而Zhong等[14]发现Co2C棱柱能够高选择性地生成低碳烯烃。其低碳烯烃的选择性能够达到60%,烯烷比高达30。他们认为助剂Na和锰对于碳化钴棱柱的形成起到了促进作用。
3.2 Fe、Co基催化剂尺寸效应
荷兰Utrecht 大学De Jong 教授在弱相互作用的碳纳米纤维(CNF)载体上通过调控负载量,用浸渍法制备了3~25nm的Co纳米粒子,利用瞬态稳态同位素动力学分析方法(SSITKA),发现费-托反应活性、转化频率(TOF)和Co 粒径间存在尺寸效应[15](图4)。在临界尺寸内,TOF随着粒径增大而增加。当Co 颗粒小于临界尺寸时,可能由于粒径越小,边角位的活性位点被CO 锁住,亦或是台阶位的数量降低。当Co颗粒大于临界尺寸时,FTS反应的TOF 不受Co 颗粒大小的影响。反应条件不同,临界尺寸大小也不同。压力为0.1MPa 时,临界尺寸为6nm;而压力为3.5MPa 时,临界尺寸为8nm。除此之外,烃类产物分布也有所改变,粒径越小,H覆盖度越高,甲烷选择性提高,但长链产物(C5+烃类)的选择性随着Co 颗粒尺寸减小而降低[16]。
De Jong 教授发现在Fe 基催化剂上也存在“尺寸效应”(图5)。同样,采用CNF为载体,利用柠檬酸铁铵为前体,用浸渍法改变Fe 的负载量控制粒径[17]。平均Fe 粒径由7.0nm 降到2.0nm,0.1MPa条件下表观TOF提高了6~8倍,甲烷和烯烃选择性没受影响。2MPa 下,在Na、S 促进的Fe 催化剂上,同样的粒径降低导致了表观TOF增加2倍,甲烷选择性显著提高。他们还假定甲烷在高活性低配位的边角位处生成,粒径的降低促进了这种活性位数量的提高。而低碳烯烃生成在台阶位,这种活性位不受粒径影响。

图4 不同反应压力下Co颗粒尺寸大小与TOF的相互关系及Co粒径对活性和甲烷选择性的影响[15-16](1bar=0.1MPa)
3.3 助剂金属的促进效应

图5 尺寸效应对Fe/CNF催化剂合成气制烯烃选择性的影响(0.1MPa,350℃,H2/CO=1,TOS=15h)及尺寸效应对Fe/CNF催化剂TOF和选择性的影响(2MPa,340℃,H2/CO=1,TOS=1h)[17]
化学助剂通常在以下方面提高活性金属催化性能:①提高还原速率;②强化活性金属中间体的成核,提高比表面积;③提高CO 吸附位数目或种类;④稳定活性相;⑤影响中间产物二次反应速率[18]。Morales 等[19]根据助剂的作用将助剂分为两类。一是结构助剂,主要是通过改变活性金属结构,调控金属与载体氧化物间相互作用,从而提高催化活性。因此,结构助剂只改变催化剂表面活性中心的数量,通常仅影响催化活性和稳定性,但不改变产品选择性。如Zr、La 等金属助剂稳定载体氧化物,避免Fe、Co 与载体形成金属氧化物。二是电子助剂。通常通过电子转移方式调变金属局域电子环境,也可影响催化剂对反应物或产物的吸脱附性质。碱金属一般通过较强的表面供电子作用,直接修饰Fe、Co 催化剂表面电子结构,影响CO、H2吸附解离、中间体脱附加氢等提高FTO 反应烯烃选择性。此外,Mn对Fe、Co都可以通过形成合金改变电子状态。
结构助剂Si、Al 和Mg 的加入可以抑制烧结,与Fe、Co 形成硅酸和铝酸盐,稳定活性相,但会抑制Fe、Co 的还原,提高机械强度[20]。此外,Zr修饰的Co/Al2O3催化剂可以阻止铝酸钴的生成[21],引入B 元素可抑制积炭、提高Co 的稳定性[22]。Büssemeier 等[23]发现Zn或V、Zn为助剂的非负载型Fe 催化剂,在320℃、1MPa、H2/CO=1 的条件下,CO转化率85%~86%,C2~C4烯烃选择性59%~71%。此后,该团队又报道了由Fe、Ti、Zn、烧结而成的氧化物催化剂在87%的合成气转化率下得到低碳烯烃选择性75%,甲烷选择性低至10%,催化剂寿命几百小时,同时伴随着大量的碳沉积[24]。Roy等[25]研究了Zn 浸渍的沉淀法Fe-Ti 催化剂,在250℃、0.25MPa、H2/CO=1 的条件下,催化剂转化率45%,C2~C3烯烃选择性68%,CH4选择性接近20%。
碱金属助剂被广泛用来提高烯烃选择性。加入碱金属(K、Na 等)后,影响了Fe 的还原,促进了碳化,导致催化剂活性、烯烃选择性和链增长能力提高,烯烃在催化剂表面上的二次加氢被抑制。而碱金属的促进作用受浓度、制备方法影响较大,通常其加入需适量。再者,碱金属修饰的催化剂也有不足,通常在反应中容易破碎,碱金属会流失等[26-28]。硫对费-托催化剂具有毒化作用,特别是Co 催化剂。但是对于Fe 催化剂,近来有报道认为适量的硫可提高Fe 催化剂低碳烯烃选择性,减少甲烷生成,低浓度添加甚至可提高催化剂活性[29-32]。Goldwasser 等[33]通 过 共 沉 淀 将K 掺 入 了LaFeO3的晶格中,制备了钙钛矿型氧化物La1-xKxFe1-yMnyO3,在280℃、1.1MPa、H2/CO=2 下得到14%CO 转化率,甲烷选择性10%,低碳烯烃选择性70%。
除此之外,Mn 既能作为结构助剂,也能起到供电子的作用。Zhang 等[34]利用超低温共沉淀法制备了FeMn 复合金属氧化物催化剂,发现Mn 降低了纳米晶体的表面能,削弱了纳米晶团聚倾向,导致随着Mn/Fe 比的提高、结晶度下降及微球晶粒尺寸的降低。此外,由于Mn 的离子半径大于Fe,结合粒径的降低共同诱导了Fe 氧化物的晶格畸变,扰乱了Fe 的晶体结构。在还原和活化后的FeMn 催化剂中,Mn 主要以MnO 的形式存在,在中低温FTO 反应中(260℃、2MPa)可能与表面吸附的CO2*中间体反应,形成MnCO3。Mn 促进了Fe2O3/(Fe,Mn)2O3向Fe3O4/(Fe,Mn)3O4的还原,稳定了Fe1-xMnxO,同时在活化和反应过程中抑制了Fe1-xMnxO 的还原和碳化。特别地,碳化物组成随着Mn 含量提高由χ-Fe5C2转变为以ε-Fe2C 为主,反映了Mn 抑制了Fe 碳化物的相转变。随着Mn 含量增加,催化活性由晶粒尺寸降低(表面积增大)以及活性相碳化铁的含量降低共同影响,呈现出火山形曲线。此外,Mn 易于在催化剂表面富集,高Mn/Fe 比可能导致催化活性的降低。FeMn(4-1)催化剂在260℃、2MPa的FTO条件下具有最高的稳态C2~C4烯烃选择性(60.6%),适量Mn的加入有利于提高烯烃产率。除了提高Fe的分散外,适量的Mn可能提高催化剂的表面碱性,对Fe 提供电子促进CO 的吸附解离,同时削弱中间体的加氢,提高烯烃选择性。此外,随着Mn含量上升,碳化物组成的变化也可能影响选择性或产物分布。孙予罕等[14]研发了一种高性能CoMn 复合氧化物催化剂,该催化剂在Mn的作用下存在显著的晶面效应,在反应过程中形成暴露(101)和(020)晶面的Co2C 纳米平行六面体。该材料在温和条件下[250℃、0.1~0.5MPa、2000mL/(g·h)、H2/CO=2],低碳烯烃选择性60.8%,甲烷低至5%,特别是C2~C4的O/P比能达到30。
3.4 金属-载体相互作用
金属载体相互作用对金属粒子得失电子能力、氧化还原能力、几何形貌和分布等都有显著的影响。载体也可以提高催化剂机械强度和热稳定性。在较为苛刻的工业费托反应条件下,载体必须具有较好的水热和机械稳定性。比表面积大的氧化硅和γ-氧化铝为常用载体,采用浸渍法以无机Fe 盐为前体即可很容易地得到高分散的纳米Fe、Co。但Fe、Co往往与氧化物载体间存在较强的相互作用,形成混合氧化物,如铝酸钴、硅酸铁等,不利于Fe、Co 的还原和活性相生成。因此,弱相互作用的载体有利于Fe 相的活化,促进助剂对Fe 的助催化作用,但制备的催化剂往往稳定性较差。
Baker等[35]用热处理的γ-氧化铝浸渍Fe和稀土金属的前体,发现在Fe-Pr/热处理γ-氧化铝催化剂上,280℃、0.8MPa、H2/CO=0.5 得到CO 转化率15%,甲烷选择性7%,低碳烯烃选择性63%。Torres等[36]采用具有弱相互作用的α-Al2O3为载体制备了Fe/α-Al2O3催化剂,Fe纳米粒子能够均匀分散在载体表面。在340℃、2MPa、H2/CO=1下,CO转化率80%,CH4选择性11%,低碳烯烃53%。
Zhang 等[37]发现对于改性多壁碳管负载纳米铁(Fe/MWCNTs)的催化体系,在FTO反应中,缺陷富集、N掺杂、O官能团化的碳管负载Fe催化剂的初始TOF、烯烃选择性、烯烃/烷烃比和链增长因子逐渐降低。在FTO 反应中(2MPa、260℃),缺陷富集的Fe/HT-CNT稳态链增长因子α值接近0.7,具有最高的C2~C4、C2~C7烯烃的选择性,分别为49.1%和72.4%。构-效关系研究表明,缺陷富集的HT-CNT 对纳米Fe 具有吸电子效应,含有吡啶、吡咯和季N 基团的N-CNT 具有给电子效应。煅烧和还原后,含氧官能团(如—OH、—OCOR、—OR、—COC—、—COOH 等)富集的O-CNT 上不稳定的羧基和酸酐等吸电子基团分解,因此表现出供电子效应。Fe/HT-CNT、Fe/N-CNT 和Fe/OCNT上催化剂的还原性和丙烯的吸附强度递增。H2还原后的Fe/MWCNTs 稳定了亚稳态FeO,在高碳势的FTO 过程中转变为活性相έ-Fe2.2C/ε-Fe2C。此外,表面缺陷对纳米Fe 存在锚定效应,促进形成了类似于活性相碳化铁的“Fe-C”配位结构,导致强CO 吸附、活化及向Fe 碳化物的快速相转变。相反,表面含O 基团抑制Fe-CNT 间相互作用并阻碍活性相的形成,但强的Fe-OCNT 相互作用提高了催化稳定性。增强的CO吸附将同时抑制H2吸附和表面中间体的加氢,与抑制烯烃再吸附和二次加氢共同促进Fe/HT-CNT上烯烃的选择生成。
3.5 双功能催化剂以及OX-ZEO催化剂
除了载体和助剂改性,采用双功能催化剂也是调控催化性能的方向之一。
一种思路是通过以费-托合成活性金属纳米Fe、Co、Ru 活化CO 进行C—C 偶联,再耦合分子筛表面Bronsted 酸位的C—C 断裂,控制链增长和适度加氢,获得目标产物低碳烯烃。日本Toyama大学Tsubaki 教授是这一理论的主要开创者,尽管该思路得到的催化剂烯烃选择性较低,但是在制备汽油组分C5~C11支链和异构烃类时表现出独特的优势[38-40]。
Kang 等[41]用Fe、Cu 的硝酸盐以及碳酸钾浸渍ZSM-5、丝光沸石、β-分子筛,在300℃、1MPa、H2/CO=2 条件下测试发现,酸性位含量最低的Fe-Cu-K/ZSM-5在81%CO转化率下,甲烷选择性20%,低碳烯烃30%。丝光沸石和β-分子筛负载的催化剂反应中结构明显被破坏。Xu 等[42]研究了K2O和MnO对Fe/Silicalite-2选择合成低碳烯烃的促进作用。在347℃、2MPa、H2/CO=2 条件下,低碳烯烃选择性最高为70%。Xu 等认为MnO 限制了C2H4和C3H6的加氢,K2O 同时提高了CO 吸附,抑制了烯烃歧化和二次加氢,共同导致了高活性(90%)和高烯烃选择性。Das 等[43]报道了Mn 的加入对Silicalite-1 负载Fe、Co 基催化剂的影响。采用Fe、Co、Mn的硝酸盐为前体,在275℃、2.1MPa、H2/CO=1条件下,CO转化率5%,10%Fe-5%Mn/Sil-1催化剂低碳烯烃选择性65%。此外,他们发现Mn加入后,铁氧化物粒径减少,碳化被抑制。
另一种思路是通过非费托活性金属的氧化物,先将合成气转化为甲醇或甲氧基中间体,再通过分子筛将中间体转化为低碳烯烃。Jiao 等[4]报道通过ZnCrOx和中孔SAPO-34 耦合,低碳烯烃的选择性和CO转化率分别达到80%和17%。Cheng等[5]报道通过ZrO2-ZnO 和SAPO-34 的耦合获得了74%的低碳烯烃选择性。他们认为ZrO2虽然可以吸附CO,但不能活化H2。Zn 以及In 的引入可以增强H2的离解,从而提高了CO的转化率。DFT研究表明,CO倾向于吸附到无空位表面附近的In—O键上,从而产生甲醇。为了提高对C2~C4烯烃的选择性,必须通过减小颗粒尺寸来减少甲醇在双金属氧化物中的停留时间。InZr的粒度越小,甲醇越容易从氧化物扩散到沸石。在将颗粒尺寸从0.85mm 减小至0.18mm 后,CO 转化率和对烯烃的选择性显著提高。
锌掺杂的ZrO2和SSZ-13 耦合则通过甲醇/DME为中间体进行合成气制低碳烯烃[44]。作者认为往ZrO2中掺杂少量的Zn 能够促进H2的杂化解离,从而为CO 的氢化提供了氢物种。双功能催化剂中的布朗斯特酸密度也起着至关重要的作用。对于CO转化率方面,在不同的酸密度下,其转化率从5%到25%不等。另外一项研究证明,对于ZnCrOx-MOR的耦合催化剂来说,乙烯酮是反应的中间体[45]。从使用129Xe 作为探针分子的固态NMR 光谱中,观察到乙烯酮可以进入8MR和10MR。同时,当使用甲醇作为进料时,12MR与8MR的比率降低,这表明甲醇更喜欢进入12MR。DFT 计算表明甲醇在12MR 上的吸附能量低于8MR。实验研究还表明,单一的乙烯酮进料对烯烃的选择性(89%)比甲醇进料高得多。
3.6 CO加氢动力学及反应机理
Albert 等[46]对CO 加氢进行了单反应中心和双反应中心模型的动力学研究。他们发现催化剂的结构除了对基本反应速率的直接影响(如CO 的解离和烃链的裂解)外,也可能间接地影响整体性能。对于单反应中心模型来说,生成C2+产物的CO 消耗速率正比于CHx插入链增长速率和链终止速率的四次方根,并且随CO 压力的增加而降低,这是因为催化剂会随着链的不断增长而中毒失活。而双反应中心模型则解决了CO 转化率随链增长下降的问题。
Zhang 等[47]对无助剂和Mn 助剂促进的Fe/SiO2模型催化剂进行了本征动力学研究。研究表明Mn加入导致烃类的生成活化能降低及CO 反应级数的下降。说明Mn 的加入促进了CO 的解离吸附和单体的形成,并且提高了表面含碳中间体的吸附。含碳物种覆盖率的增加有利于含碳物种的偶联,同时抑制甲烷化和碳链终止反应。此外,烯烃的生成活化能下降显著,说明Mn 助剂有利于烯烃生成。H2对烷烃反应级数为正,对烯烃和CO2为负,说明催化剂表面H 对于烯烃生成已经足够,H2分压的提高不利于烯烃生成。在FeMn 催化剂上对于烯烃生成,CO 和H2级数同时降低,说明CO 和H2可能吸附在不同的活性位上。Mn 的加入导致CO2生成的活化能提高,强化的CO 解离吸附减少了分子吸附的CO 并且抑制了表面上的C*和O*的重新结合,此外,少量的Mn可以通过促进Fe3O4的还原和碳化来抑制水气变换反应。反应过程中,在FeMn 催化剂表面大量覆盖强吸附的CO 和解离的含C 物种,阻碍了H2的吸附解离,同时削弱了烯烃再吸附和二次加氢。上述促进作用共同降低了CH4和CO2的选择性,最终促进了低碳烯烃选择性提高。
在Fe、Co基催化剂上,CO加氢反应是表面聚合反应,机理非常复杂。目前学术界提出的主要有3 种机理(表2),假定了3 种不同的链增长单体[48-49]。尽管费-托合成发现已经95 年,但其机理仍存争议。为了进一步理解反应机理,需要应用多学科的方法,如模型表面科学实验[50-51]、密度泛函理论计算、宏观动力学研究等来进行验证。从费-托合成机理上看,对于合成气直接制烯烃过程,以下3 个中间过程对于烯烃选择性有较大影响。①水气变换和Boudouard 反应[52]:水气变换直接影响CO 转化的效率,降低水气变换,让O 以水的形式脱除,可以有效提高烯烃的时空收率。Boudouard 反应在高温下是热力学有利的。②碳碳偶联:作为费-托合成中最典型、最重要的反应,直接决定产物分布和组成,由于费-托反应的聚合本质,这一过程被认为最不可控。提高FTO 反应性能,很重要一点是控制产物分布和链增长范围。③烯烃再吸附和二次反应:烯烃二次反应主要包含二次加氢、再插入、异构化和裂解等,作为副反应,直接影响烯烃选择性。
Iglesia 等[53]对CO 和H2在催化剂表面的活化和解离做了系统的动力学实验和理论计算,提出CO的活化主要通过两种机理:①CO 在表面直接解离形成C*和O*物种;②加氢辅助的C—O 键断裂及转化,如图6所示。在Fe催化剂上,直接解离和H协助CO*解离同时存在,两种方式都能生成CH2*单体,区别在于O 的脱除路径。相比而言,O 以H2O 形式脱除,即H 协助CO*解离更占优势。通常,高反应温度或碱金属助剂会显著促进CO 直接解离。在Co 催化剂上,非解离CO*不如H 协助解离容易,H 协助CO 解离是主要的活化路径,因此O主要以水的形式脱除。

表2 费-托合成反应机理[48-49]

图6 费-托合成中CO活化的基元步骤[53]
4 总结与展望
合成气是重要的能源转化平台,合成气直接制烯烃过程是实现煤、天然气、生物质等能源清洁高效利用的有效途径,有利于实现国民经济的可持续发展,确保能源战略安全,具有潜在的工业应用价值。合成气直接制烯烃反应的目标是提高催化效率、目标产物烯烃选择性及催化剂稳定性,这要求催化剂具有分子剪切能力,抑制甲烷、长链烃类、CO2等副产物的生成能力。对于传统的Fe、Co基催化剂,提高合成气制烯烃的关键问题是控制反应物CO 在活性相表面吸附、解离和加氢。通过改变载体或修饰载体表界面调控金属-载体相互作用;通过引入助剂调控助剂促进作用或合金效应;通过先进制备方法调控活性金属自身粒径、暴露晶面或形貌;通过改变催化环境,如气氛诱导等可以有效地对催化剂的性能进行调控。
能源是人类社会永恒的话题,全球经济尚处高速发展中,对能源的需求与日俱增,但国际能源供应常受国际形势的剧烈影响。低碳烯烃(乙烯、丙烯、丙烯)是各种大宗化学品的原料,随着世界经济的复苏,未来对烯烃的需求将长期保持较快的增速。合成气直接制低碳烯烃,无疑对于清洁高效利用非石油资源意义重大。
但合成气直接制低碳烯烃工艺仍面临许多问题。
(1)基础研究
①反应机理方面 由于费托合成反应复杂,中间产物繁多,反应机理很难理清。研究结论常涉及包容性问题以及框架效应。包容性问题在于给定理论的题设和结论的简洁性和科学性。框架效应在于不同科研人员即使针对同一问题,研究手段和侧重点各不相同,进而造成了既得理论的差异。
②催化剂设计方面 一般认为费托合成产物链长选择性(只是链长,不区分烯烃还是烷烃)受到ASF分布的限制。突破ASF分布的关键在于打破其核心假设,即不同碳数的反应中间体链增长能力相同。同时要在控制链长的基础上还要保证高烯烷比。因此,目前对于催化剂设计的研究关键在于发掘既能控制链增长能力又能控制烯烷比的催化剂。
(2)工业应用
真实工业反应条件要比实验室条件更为复杂。就催化剂而言,要考虑催化剂的抗毒性和水热稳定性。而往往性能可观的催化剂在负载后性能会大幅下降,这是一个核心问题。对于反应器设计,要求反应系统良好的导热性,要防止局部过热导致催化剂失活。
(3)产业联合
抛开战略性,就经济性而言,合成气制低碳烯烃要形成规模经济才能盈利。这个规模经济包括自身的工艺规模以及与上下游的联合产业规模。要对整个生产系统进行优化,形成产业联合,才有可能实现规模经济,形成成本优势,进而推动技术优势,从而获取整个产业的比较优势。
