Transcriptome and Metabolome Analyses of Sea Cucumbers Apostichopus japonicus in Southern China During the Summer Aestivation Period
2021-03-06YANGQiuhuaZHANGXushengLUZhenHUANGRuifangTRANNgocTuanWUJianshaoYANGFuyuanGEHuiZHONGChenhuiSUNQianZHOUChenandLINQi
YANG Qiuhua , ZHANG Xusheng , LU Zhen HUANG RuifangTRAN Ngoc Tuan WU Jianshao YANG Fuyuan GE HuiZHONG Chenhui SUN Qian ZHOU Chen and LIN Qi
1) Key Laboratory of Cultivation and High-Value Utilization of Marine Organisms in Fujian Province, Fisheries Research Institute of Fujian, Xiamen 361013, China
2) Guangdong Provincial Key Laboratory of Marine Biology, Marine Biology Institute, Shantou University, Shantou 515063, China
Abstract Aestivation is a common strategy of sea cucumbers (Apostichopus japonicus) in response to high-temperature conditions.Previous studies have individually investigated the immune and physiological alterations at the aestivation stage. However, these studies have not evaluated the relationship between immunity and physiology. In this study, we explored the transcriptome and metabolome of A. japonicus during the aestivation stage to study the relationship. The transcriptome analysis of dormant (aestivation)and revived A. japonicus generated 2368 differentially expressed genes, including 927 downregulated genes and 1441 upregulated genes. Based on Gene Ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analyses, the downregulated genes in the dormant group were found to be involved in DNA replication, RNA metabolic process, and protein metabolism, which results in the inhibition of motility, skeletal development, neural activity, cell proliferation, and development of A. japonicus. In contrast, the upregulated genes were found to be associated with fatty acid metabolism, carbohydrate hydrolysis, and phagocytosis. In the metabolome analysis, the downregulated metabolites were found to be associated with fatty acid metabolism, starch and sucrose metabolism, and TCA cycle. This indicates that dormant sea cucumbers consume reserved carbohydrates and fatty acids to maintain low levels of energy supply. The protein-protein interaction network analysis further revealed that carbohydrate hydrolysis promoted phagocytosis activity in the dormant group. This study provides new insights into potential molecular mechanisms of sea cucumber survival in high-temperature conditions, which is critical in aquaculture of sea cucumbers.
Key words aestivation; Apostichopus japonicus; hypometabolism; fatty acid metabolism; carbohydrate hydrolysis; phagocytosis
1 Introduction
Sea cucumber (Apostichopus japonicus) is an echinoderm belonging to the Holothuroidea family (phylum Echinodermata). It is well known that several species of sea cucumbers are detritus feeders, and this enables them to ingest sediments such as organic matter, bacteria, and protozoa to acquire nutrients. Therefore,A. japonicusis very important for maintaining a clean environment (Yanget al.,2006). The body wall ofA. japonicusis abundant in collagen and mucopolysaccharides and is highly valuable in medicine and commerce (Jiet al., 2008; Duanet al., 2010),which explains why sea cucumbers are widely cultured in many countries, including Japan, Korea, and China (Zhaoet al., 2014).
In China,A. japonicusis an important commercial mariculture species, and 174340 metric tons (including 29829 metric tons in Fujian province) ofA. japonicuswere produced in 2018 (Weiet al., 2015; DOF, 2019). Most previous studies focused only on the distribution of this species in northern China (Zhaoet al., 2014; Liet al., 2019).In recent years, sea cucumbers have been popularly cultured in southern China (Yuet al., 2014; Fanget al., 2017).Previous studies showed that different culture environments significantly affected the metabolism and immunity of sea cucumbers (Wuet al., 2009; Gonzάlez-Wangüemertet al., 2018). Therefore, the metabolism and immune responses ofA. japonicuscultured in southern China may differ from those ofA. japonicuscultured in northern China, which can be attributed to the different climate conditions between the two regions. Because the temperature in southern China is usually high, it is important to understand the effect of high temperature on the development ofA. japonicuscultured in the coastal waters in south of China. Aestivation is a common strategy in both vertebrates and invertebrates for survival under high-temperature and arid conditions (Storeyet al., 2012). Sea cucumbers have been found to enter an aestivation state when the environmental temperature increased, especially during summer, with temperatures over 25℃ (Zhaoet al.,2014). Furthermore, temperatures over 20℃ provide a favorable condition for multiplication of microorganisms,which culminates in a large-scale outbreak of diseases that severely threaten the survival of multicellular organisms(Lacosteet al., 2001; Elliotet al., 2002; Altizeret al., 2013;Lokmeret al., 2015). Therefore, aestivation of sea cucumber is a protective strategy for resisting to pathogens through upregulating the expression of immunity-related genes, including lysozyme, C-type lectin, and complement factors(Wattset al., 2012; Zhaoet al., 2014; Liet al., 2019). Although metabolism and immunity of dormant sea cucumbers have been investigated respectively in previous studies, the relationship between their metabolism and immunity needs further studies (Wanget al., 2008; Yuet al.,2014; Zhaoet al., 2014; Liet al., 2019). In this study,transcriptome sequencing was used to study the molecular responses ofA. japonicusduring the aestivation stage in southern China.
As demonstrated in many studies, high-throughput RNAsequencing (RNA-seq) technology is an excellent approach to study the transcriptional level of large numbers of genes in tissues or organisms. The sequencing results can provide a ‘whole picture’ of the gene expression profile in an organism (Duet al., 2012; Huanget al., 2016;Liet al., 2019; Zhanget al., 2019). In addition, mass spectrometry-based metabolome study is a new approach that provides a better understanding of the whole organism,enabling the measurement of all small molecules, chemicals, and metabolites in a given sample, which further validates and explains the transcriptome sequencing analysis (Nicholsonet al., 2008; Stringeret al., 2016). Therefore, our study employed transcriptome sequencing and metabolome analysis to investigate the alteration of physiological metabolism and immune responses in the aestivation stage, and analyzed the potential relations between them. The results of this study will provide new insight into the potential molecular mechanisms of physiological metabolism and immune responses, which are crucial for the survival of sea cucumbers in aquaculture.
2 Materials and Methods
2.1 Sample Collection
HealthyA. japonicusindividuals (30 – 50 g) were acquired from Dongshan base of Fujian Fisheries Research Institution (Zhangzhou, Fujian, China) and acclimatized in seawater (pH: 7.9 – 8.3; salinity: 30 – 32) at Fujian Fisheries Research Institution (24˚29΄6.92΄΄N, 118˚04΄32.49΄΄E).The formula feed (Jinpai Inc., Qingdao, China) was mixed with sea mud at a ratio of 1:4 (w/w) and fed to sea cucumbers twice daily (06:00 and 18:00) until aestivation.The tissue mixture (including 3 – 5 g muscle, 0.5 – 1 g intestine, 0.5 – 1 g intestinal tissues, and 1 mL coelomic fluid) represented the whole body of sea cucumber. Tissue mixtures from three sea cucumbers were mixed together (approximately 20 g) and considered as one experimental sample for minimizing individual deviation. In 2018, the temperature of the culture water was over 25℃ from May to September, which induced the entry ofA. japonicusinto the aestivation state (Fig.1). In addition, the average temperature was 24.4℃, making the sea cucumbers dormant in October. In contrast, the temperature in January, February, and March was 15.3℃, 14.7℃, and 16.4℃, respectively, which revived the sea cucumbers from aestivation(Fig.1). Thus, samples collected from May to October and from January to March were used to investigate the aestivation and revival ofA. japonicus, respectively. Samples collected in May, June, July, August, September, and October were named dormant group (DG), whereas samples collected in January, February, and March were named revival group (RG). After collection, the samples were immediately stored in liquid nitrogen for extraction of RNA and metabolites.

Fig.1 Temperature curve of 2018.
2.2 cDNA Library Construction
A total of 10 g tissue mixture (from three individuals)was ground using a ceramic mortar and pestle. Total RNA was extracted using the Trizol Kit (Invitrogen, USA) according to the manufacturer’s instructions. The quality and quantity of total RNA were estimated using 1% gel electrophoresis, Nano Photometer spectrophotometer (IMPLEN,CA, USA), and Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). The total RNA obtained was used to synthesize cDNA libraries using PrimeScript RT reagent Kit with gDNA Eraser (Takara, Dalian, China) according to the manufacturer’s instructions. The cDNA was used as a template for quantitative real-time PCR (RTqPCR) and RNA-seq transcriptome analyses.
2.3 RNA-seq
At least 1 μg of total mRNA was used to construct cDNA libraries. For the construction of the cDNA library, purified mRNA from total RNA was cut into fragments using NEBNext®UltraTMRNA Library Prep Kit (Illumina, USA).Briefly, mRNA was purified from total RNA using oligo(dT) magnetic beads. NEB fragmentation buffer was used to randomly cut mRNA into small fragments, which were then used as templates for synthesizing the first-strand cDNA using random hexamer primer and M-MuLV Reverse Transcriptase. Subsequently, DNA Polymerase I and RNase H were used to produce second-strand cDNA using dNTPs.Double-strand cDNA was purified, end-repaired, adenylated (A-base addition), and ligated of the Illumina-idexed adaptors. Finally, 150–200 bp cDNA was used for PCR amplification, and PCR products were purified using AMPure XP beads (Beckman Coulter, Beverly, USA), which was used to generate the cDNA libraries. The cDNA library quality was further assessed using the Agilent Bioanalyzer 2100 system. A concentration of cDNA of > 2 nmol L−1was used for sequencing with the Illumina HiseqTM2500 platform. All original sequence data in this study were submitted in NCBI Sequence Read Archive database(https://www.ncbi.nlm.nih.gov/bioproject/PRJNA596892)under accession number PRJNA596892.
2.4 Sequence Assembly and Gene Function Annotation
Quality control was performed to remove adaptor sequences and poly-N-containing and low-quality reads.Thereafter, Q20 and Q30 of clean reads were calculated.HISAT2 version 2.0.5 software was used to further align and obtain the clean reads to reference the genome ofA.japonicus, with the accession number PRJNA354676 (Zhanget al., 2017). Assembly of the novel transcript was performed using StringTie version 1.3.3b, and these transcripts were annotated based on Protein family (Pfam), Superfamily,Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) database. FeatureCounts version 1.5.0-p3 was used to count the number of genes. Based on the gene length and read count, fragments per kilobase of transcript per million mapped reads (FPKM) of each gene was calculated. The subsequent fold change calculated was based on the FPKM ratio between DG and RG. DESeq2 was used to identify genes with aP-value of < 0.05 and log2(Fold change) of > 1 as differentially expressed genes(DEGs). Obtained genes were annotated using Pfam, Superfamily, GO, and KEGG databases for characterizing their functions at both molecular and cellular levels (Kanehisaet al., 2000; Gene Ontology Consortium, 2004; Finnet al., 2014; Liet al., 2018).
GO enrichment of DEGs was performed using cluster-Profiler R package, and the bias of gene length was corrected. For a better understanding of gene functions in a biological system, KEGG enrichment was performed using clusterProfiler R package, which clearly explained the genes at molecular levels (Gene Ontology Consortium,2004; Liet al., 2018). Furthermore, Venn diagram analysis was used to examine the special expression of genes in each group, and GO and KEGG enrichments were used to analyze the functional categories (Liet al., 2018; Liet al.,2019).
2.5 Protein-Protein Interaction (PPI) Analysis
PPI analysis of DEGs was carried out using the STRING database (http://string-db.org/), which investigates the direct and indirect associations between different proteins(including stable complexes, metabolic pathways, and a large array of direct and indirect regulatory interactions)(Szklarczyket al., 2011; Liet al., 2019). Networks showing the up- and downregulated genes were constructed using Cytoscape software based on the results of STRING analysis.
2.6 LC-MS Analysis
A total of 10 g tissue mixture (from three individuals)was grounded with liquid nitrogen and resuspended with 80% methanol and rigorously vortexed. The samples were incubated at −20℃ for 60 min and then centrifuged at 14000×gat 4℃ for 20 min. The supernatants were subsequently transferred into a fresh tube and spun in a vacuum concentrator until dry. The dried metabolite pellets were reconstituted with 60% methanol and analyzed using LC-MS/MS. The supernatant was loaded into an Agilent 1290 Infinity LC System (Agilent Technologies, Germany) and ACQUITY UPLC BEH Amide reverse-phase column (2.1 mm × 100 mm × 1.7 μm) (Waters, USA). After loading, the sample was filtered and centrifuged to remove particles. A mixture of 25 mmol L−1acetic acid and 25 mmol L−1ammonia water and acetonitrile were used in the mobile phases: A and B, respectively. Elution was performed with 5% A and 95% B for 0.5 min, 35% A and 65% B for 6.5 min, and 60% A and 40% B for 2 min, and then eluted with 5% A and 95% B for 3.5 min to balance the column.The total chromatographic elution process lasted 12 min and the flow rate was 500 μL min−1.
Raw data generated by UHPLC-MS/MS were processed using Compound Discoverer 3.0 (CD3.0, Thermo Fisher)for peak alignment, peak picking, and quantification of each metabolite. The main parameters were set as follows: retention time tolerance = 0.2 min; actual mass tolerance = 5 ppm; signal intensity tolerance = 30%; signal/noise ratio =3; and minimum intensity = 100000. Thereafter, peak intensities were normalized to the total spectral intensity. The normalized data was used to predict the molecular formula based on additive ions, molecular ion peaks, and fragment ions. Peaks were matched with mzCloud (https://www.mzcloud.org/) and ChemSpider (http://www.chemspider.com/) databases to obtain accurate qualitative and relative quantitative results. The different metabolites obtained were identified based on the following principles: theoretical fragments should exhibit three strong peaks that match the secondary characteristic fragments and should cover more than 80% of the secondary characteristic fragments.Significant differences (P <0.05) in terms of each variable were first detected using thet-test; metabolites (P <0.05)related to fatty acid metabolism, carbohydrate metabolism,and TCA cycle were selected for subsequent analyses.
2.7 Quantitative Real-Time PCR (RT-qPCR)Analysis of Metabolism-Related Genes
To validate the gene expression results obtained from the transcriptome sequencing, the tissue mixture of six individuals were sampled in August (highest temperature)and February (lowest temperature) and were used to detect gene expression using SYBR®Premix Ex TaqTMII Kit(Takara, Dalian, China) in LightCycler®480 (Roche, USA).Each group contained six replicates. Total RNA fromA.japonicuswas extracted using Total RNA kits Ⅱ (Omega,USA) according to the manufacturer’s instructions. cDNA was synthesized using PrimeScript RT reagent Kit with gDNA Eraser (Takara, Dalian, China) according to the manufacturer’s instructions. The obtained cDNA was used as a template for RT-qPCR. In this study, 18 genes relating to carbohydrate metabolism, fatty acid metabolism, TCA cycle, and phagocytosis pathways were analyzed with RTqPCR. A 20-μL reaction mixture contained 10 μL of SYBR®Premix Ex TaqTMII, 2 μL of cDNA, 0.8 μL (10 mmol L−1) of each (forward and reverse) primer (Table 1), and 6.4 μL of ddH2O. The amplification was programmed as follows:denaturation at 95℃ for 30 s, followed by 40 cycles of 95℃ for 5 s and 60℃ for 20 s; melting curve was analyzed from 65℃ to 95℃. Each sample was analyzed in triplicate. The relative expression of selected genes was calculated by normalizing it to the internal control gene(β-actin) through the 2−ΔΔCtalgorithm method. All experiments were performed with three replicates and data of RT-qPCR analysis were analyzed using one-way analysis of variance (ANOVA).

Table 1 Primer sequences used in this study
3 Results
3.1 Transcriptome Quantification
A total of 474876912 raw reads were acquired after RNAseq. Of these raw reads, a total of 457223622 clean reads(96.28% of the raw reads) were obtained after removing the adaptor sequences, poly-N-containing reads, and lowquality reads (Table 2). Then 301336131 reads (65.90% of clean reads) were mapped to the reference genome ofA.japonicus,which was composed of 279721413 (61.18%of clean reads) uniquely mapped reads and 21614718(4.73% the clean reads) multiple mapped reads. The average Q20 and Q30 of the read data was 97.43% and 92.90%,respectively. The QC content accounted for 41.18% of the sequencing data.
3.2 Characterization of DEGs
A total of 2368 DEGs (including 927 downregulated and 1441 upregulated genes) and 39081 unchanged genes were observed in DG compared with that in RG (Fig.2A).Hierarchical clustering analysis of DEGs was performed to identify group clusters. The results showed that DG(samples in May, June, July, August, September, and October) and RG (samples in January, February, and March)were divided into two clusters (Fig.2B).
GO and KEGG analyses were used to further characterize the function of these DEGs. In DG, GO analysis revealed that the down- and upregulated genes were assigned to sub-categories under three primary categories,including biological process, cellular component, and molecular function (as shown in Fig.3). The downregulated genes were assigned to 41 sub-categories, which were mainly classified into DNA replication, RNA metabolic process, protein modification and biosynthesis, macromolecule metabolism, and cellular metabolism (Fig.3A). In contrast, upregulated genes were assigned to 85 sub-categories, such as peptidase activity acting on L-amino acid peptides, peptidase activity, proteolysis, endopeptidase activity, metallopeptidase activity, serine-type endopeptidase activity, serine-type peptidase activity, and serine hydrolase activity (all belonging to the protein degradation category),etc. In addition, the carbohydrate metabolic process was also significantly enriched in DG, as shown in Fig.3B.
To better understand the function of DEGs in DG,KEGG enrichment analysis was performed (Fig.4). The results showed that downregulated genes related to the gene expression process (i.e., spliceosomes, RNA transport, ribosome biogenesis in eukaryotes, and ribosomes)and proteasome metabolism pathways were significantly enriched (Fig.4A). The upregulated genes were identified to be associated with pathways related to phagocytosis(lysosomes, endocytosis, and phagosomes), lipid metabolism (fatty acid metabolism, steroid biosynthesis, ether lipid metabolism, and glycerophospholipid metabolism), and amino acid metabolism (glycine, serine, threonine, arginine, proline, tyrosine, and histidine metabolism) (Fig.4B).In addition, metabolisms related to carbohydrates (including starch and sucrose metabolism) were also enhanced in DG (Fig.4B).

Table 2 The transcriptome summary of A. japonicus
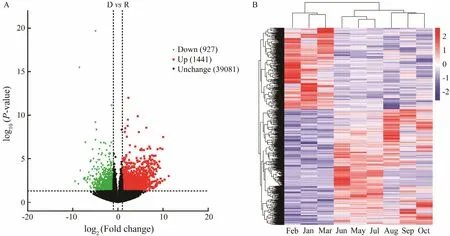
Fig.2 Volcano plot (A) and heatmap (B) of differentially expressed genes (DEGs) between the dormant group (DG) and revival group (RG). (A) X-axis exhibits log2 (Fold change) of DG vs. RG, and the Y-axis shows −log10 (P-value). Red(upregulation) and green (downregulation) dots indicate significantly different expression (P < 0.05, log2 (Fold change) >1), and black dots show no significant difference. (B) Hierarchical heatmap of all expressed genes based on the centered and normalized log10 (FKPM+1). Red color indicates high expression value, while blue color indicates low expression value.

Fig.3 Gene Ontology (GO) annotated for differentially expressed genes (DEGs) at the levels of biological process (BP),cellular component (CC), and molecular function (MF). (A) and (B) represent downregulated and upregulated genes, respectively, in the dormant group.

Fig.4 KEGG annotation of differentially expressed genes (DEGs) between the dormant group (DG) and revival group(RG). (A) and (B) represent downregulated and upregulated genes, respectively, in the dormant group.
3.3 Specially Expressed Genes Between DG and RG
In the Venn diagram analysis, 1850 and 405 genes were only expressed in DG and RG, respectively (Fig.5). These genes were further analyzed using the KEGG database.The pathways involving carbohydrate metabolism (starch and sucrose metabolism and other glycan degradation),fatty acid-related metabolism (fatty acid metabolism, biosynthesis of unsaturated fatty acids, and fatty acid elongation), and phagocytosis (phagosomes, lysosomes, and endocytosis) were found to be significantly enriched in DG compared with those in RG (Fig.6).

Fig.5 Venn diagram (A) and hierarchical clustering analysis (B) showing the number of differentially expressed genes(DEGs) between the dormant group (DG) and revival group (RG).

Fig.6 KEGG annotation of exclusively expressed genes in the dormant group (DG) and revival group (RG).
3.4 PPI Analysis of Up- and Downregulated Genes
The STRING database was used to analyze interactions among DEGs and the results of haracterization of genes in the PPI diagram. The core downregulated genes, such as methyltransferase NSUN6 (gene 100), ribosomal protein(gene 1012), translation initiation factor (gene 10825), ribosome production factor 1 (gene 13978), RNA cytidine acetyltransferase (gene 14575), U3 small nucleolar ribonucleoprotein protein (gene 16258), pre-rRNA-processing protein TSR1-like (gene 16962), eukaryotic translation initiation factor 3 subunit B (gene 17585), GMP synthase (gene 26215), and eukaryotic translation initiation factor 6 (novel 11563), were classified into transcription and translation (Fig.7A), while other downregulated genes involved in decreasing motility (dystrophin-like protein (gene 23040),ubiquitin carboxyl-terminal hydrolase 19 (gene 266), zinc finger MYND domain-containing protein 10 (gene 8809)),skeletal development (synaptotagmin-7 (novel.13845) and twisted gastrulation protein (gene 10484)), neural activity(vesicular glutamate transporter 1 (novel.6879), and choline O-acetyltransferase (gene 20445)), cell proliferation(Baculoviral IAP repeat-containing protein 2 (gene 17067)),and development (intraflagellar transport protein 27 (gene 20253), and SPARC-related modular calcium-binding protein 1 (gene 6491)) were placed in DG.
In contrast, interactions among the upregulated genes were mainly found to be related with carbohydrate hydrolysis (maltase-glucoamylase (MGAM) (gene 1069), hexokinase-1 (HK1) (gene 7968), pancreatic alpha-amylase (PAMY)(gene 2722), alpha-amylase B (AMYB) (gene 2721), alphaamylase (AMY) (gene 10557)) and promotion of phagosome(V-type H+-transporting ATPase subunit A (VATPA) (gene 25464)) (Fig.7B). These metabolic pathways that upregulated genes in the PPI analysis were also consistent with KEGG enrichment of DEGs and Venn diagram analysis.

Fig.7 PPI network of differentially expressed genes (DEGs) based on STRING analysis results. (A) and (B) represent downregulated and upregulated genes, respectively, in the dormant group. The size and color of nodes indicate connectivity.
3.5 Pathway Analysis

Fig.8 Genes involved in carbohydrate metabolism, fatty acid metabolism, TCA cycle, and phagocytosis pathways in dormant A. japonicus and their RNA-seq expression values (in TPM; transcripts per million). The black solid arrow represents direct conversion between two substances. The black dotted arrow represents indirect conversion between two substances; however, some intermediate products were omitted. The green dotted line represents interactions between two proteins.

Table 3 DEGs involved in carbohydrate metabolism, fatty acid metabolism, TCA cycle, and phagocytosis pathways in dormant A. japonicus
The pathways found in DG were elucidated in detail in Fig.8 (including genes and metabolite). The functional characterization of gene expression (transcripts per million [TPM]) and metabolites are shown in Table 3 and Table 4, respectively. In DG, high expression of genes include fatty acid synthase (FASN, 2.86-fold), fatty acid synthaselike (FASNL, 3.15-fold), very long-chain specific acyl-CoA dehydrogenase (ACADVL, 1.13-fold), long-chain specific acyl-CoA dehydrogenase (ACADL, 2.83-fold), and malonyl-CoA-acyl carrier protein transacylase (MCAT, 1.23-fold) related to pathways of fatty acid biosynthesis and degradation. The genesHK1(1.28-fold),PAMY(7.90-fold),AMYB(over 10-fold),AMY(2.86-fold), andMGAM(6.33-fold), which are related to carbohydrate degradation, were highly expressed in DG. Carbohydrate degradation also led to the production of acetyl-CoA in the TCA cycle.Based on the PPI analysis, the upregulation ofPAMY,AMYB, andAMYstimulated the expression ofMGAMandHK1. The expression ofHK1bond toUBCV, which enhanced phagocytosis throughVATPA(2.06-fold). Furthermore, some phagocytosis-related genes that were associated with pathogen scavenging were upregulated in DG.Such genes include actin-related protein 2/3 complex subunit 5A (ARPC5A, 1.46-fold), actin-related protein 2/3 complex subunit 5B (ARPC5B, 2.31-fold), PH and SEC7 domain-containing protein (PSD, 1.11-fold), N-acetylglucosamine-6-sulfatase (GNS,1.18-fold), N-acetylgalactosamine-6-sulfatase-like (GNSL, 2.99-fold), and macrophage mannose receptor 1 (MRC1, 3.30-fold).
Metabolism analysis revealed that there are 604 downregulated and 51 upregulated metabolites in DG compared with those in RG. In DG, the high expression ofMGAMreduced the sucrose level (2.16-fold) (Fig.8, Table 4). Moreover, the low level of hexadecanoic acid reduced the production of acetyl-CoA (2.4-fold), which is a substrate in the TCA cycle. Therefore, lower levels of oxoglutaric acid(2.88-fold), succinic acid (2.32-fold), and citric acid (3.85-fold) were also observed in DG.

Table 4 Metabolites involved in carbohydrate metabolism, fatty acid metabolism, TCA cycle, and phagocytosis pathways in the dormant group compared with those in the revival group
3.6 RT-qPCR Analysis
Eighteen genes related to the pathways shown in Fig.8 were validated using RT-qPCR (Fig.9). The results showed that with the exception ofFASNL, which was slightly downregulated, all tested genes related to phagocytosis, fatty acid metabolism, and carbohydrate hydrolysis were increased in DG. The relative expression of genes demonstrated that 17/18 (94%) genes showed a similar trend compared with the results in RNA-seq. These results indicate that the data obtained from RNA-seq is highly reliable.Based on RNA-seq,AMYBwas detected in DG but not in RG. The TPM ratio (DG:RG) was infinite, which is difficult to illustrate in graphics. Thus, the largest one of ten TPM values was used to replace infinity in order to show the high expression ofAMYBin DG (Fig.9).

Fig.9 Eighteen genes of the pathway analysis shown in Fig.7 were validated by comparison of RNA-Seq (transcripts per million) and qRT-PCR.
4 Discussion
The increase in summer temperature facilitates disease outbreaks caused by microorganisms, which threatens the survival of marine organisms (Lacosteet al., 2001; Altizeret al., 2013). The effects of higher temperatures on animals living in lower latitudes are usually serious (Lokmeret al., 2015). To protect against pathogen infections, sea cucumbers have evolved an aestivation behavior in response to high temperature (Duet al., 2012; Zhaoet al.,2014; Liet al., 2019). As previously stated, most studies onA. japonicuswere conducted in northern China (Anet al., 2007; Wanget al., 2008; Zhaoet al., 2014; Liet al.,2019). However, changes in metabolism and immune responses of dormantA. japonicusfor adapting to climate warming have been scarcely reported. In this study, transcriptome analysis was performed to investigate the gene expression ofA. japonicusin Fujian province with a warm climate, which is important to determine the adaption mechanism ofA. japonicusin response to higher temperatures.
Overall, 2368 DEGs (downregulated genes: 927; upregulated genes: 1441) were found in DG compared with those in RG. KEGG enrichment analysis of downregulated genes revealed that 41 GO terms and 20 KEGG pathways were found to be associated with DNA, RNA, protein, and cellular metabolisms in DG. These results imply that the physiological functions were decreased in DG, leading to inhibition of growth and development ofA. japonicusduring the aestivation period. These results are similar to those of previous studies, in which organisms reduced metabolisms of DNA, RNA, and protein during their dormant period, which further affected their growth and development(Zhaoet al., 2014; Zhuet al., 2015; Feiet al., 2016). In contrast, upregulated genes related to protein degradation,phagocytosis, carbohydrate metabolism, and fatty acid metabolism were identified through GO and KEGG analyses.Based on KEGG analysis, long-chain fatty acids and saccharides are able to produce acetyl-CoA, which plays a crucial role in ATP synthesis. Previous studies on sea cucumbers cultured in northern China also showed similar results. More carbohydrate catabolism and fatty acid degradation occur to supply energy to the sea cucumbers during aestivation (Duet al., 2012; Zhaoet al., 2014; Maet al.,2015; Feiet al., 2016). These results indicate that dormant animals maintain energy supplyviaincreasing consumption of stored carbohydrates and fatty acids during food shortage (Storeyet al., 2012). Moreover, phagocytosis-related pathways (i.e., endocytosis, lysosomes, and phagosomes) were upregulated in dormant sea cucumbers, which may play an important role in the innate immunity as a primary defense in invertebrates (Horwitz, 1984; Maliket al.,2000; Garinet al., 2001; Yateset al., 2005; Yamadaet al.,2007; Wanget al., 2013). Among these phagocytosis-related genes,MRC1was significantly upregulated (3.3-fold)in DG. In previous study, the upregulation of the lysosomes and C-type lectin was observed in dormant sea cucumbers (Zhaoet al., 2014). In the Venn diagram analysis,2255 genes expressions were affected. KEGG enrichment of these genes revealed that phagocytosis (i.e., endocytosis, lysosomes, and phagosomes) and the metabolism of fatty acid and carbohydrate were affected more significantly in DG than those in RG. Therefore, during aestivation, hypometabolism occurred, while fatty acid metabolism, carbohydrate metabolism, and phagocytosis activity increased during the aestivation stage in sea cucumbers.
In the subsequent PPI analysis, in dormant sea cucumbers, the core downregulated genes were assigned to transcriptional and translational regulations (Haaqet al., 2015).Such genes include methyltransferase NSUN6, ribosomal protein, translation initiation factor, ribosome production factor 1, RNA cytidine acetyltransferase, U3 small nucleolar ribonucleoprotein protein, pre-rRNA-processing protein TSR1-like, eukaryotic translation initiation factor 3 subunit B, GMP synthase, and eukaryotic translation initiation factor 6. Previously, decreased transcriptional level and translational level of gene expression were also observed during the dormant stage in northern sea cucumber and other animals (Zhaoet al., 2014; Feiet al., 2016; Sunet al., 2018). Once the animals enter the aestivation or hibernation states, slow movements decrease their opportunity to acquire adequate food. Food shortages cause energy deficiency in the body, so the animals reduce their levels of metabolisms and energy consumption, leading to the inhibition of most gene expression and protein translation activities (Storeyet al., 2004). Furthermore, these downregulated genes regulate the motility, skeletal development, neural activity, cell proliferation, and development ofA. japonicus, according to UniProt database analysis (UniProt Consortium, 2014). Our findings are consistent with those of other studies and suggest that sea cucumbers reduce the expression of transcriptional and translational genes related to hypometabolism, which can cosequently lead to decreased motility, stopped skeletal development, declined neural activity, and reduced cell proliferation and development (Storeyet al., 2004; Zhaoet al.,2014; Sunet al., 2018). This mechanism is in accordance with the reduction of energy consumption in response to the absence of energy intake during the aestivation state of sea cucumbers at high temperatures (Jiet al., 2008; Sunet al., 2018).
However, PPI analysis showed that upregulated genes in the dormant sea cucumbers involve carbohydrate hydrolase includingPAMY,AMYB,AMY,MGAMandHK1(Griffinet al., 1989; Maarelet al., 2002; Konget al., 2003;Simet al., 2008). Based on the STRING database,HK1interacts with ubiquitin C variant (UBCV), which improved the phagosome activityviaincreasing the expression ofVATPA, an ATP-dependent proton pump responsible for the acidification of phagosome (Ciprianoet al., 2008).These results suggest that the upregulation of carbohydrate hydrolase can promote phagocytosis in dormantA.japonicus. In addition, expressions of other phagocytosisrelated genes (ARPC5A,ARPC5B,PSD,GNS,GNSL,andMRC1) were also increased in dormantA. japonicus. Similar to previous studies, immune cells recognize pathogensviapattern recognition receptors in combination with pathogen-associated molecular patterns (Chuet al., 2013).MRC1is a typical pattern recognition receptor that ligates the mannose-rich glycoconjugates of pathogens (Ezekowitzet al., 1990; Marttilaet al., 2008). The upregulation ofMRC1indicates that the immune cells improved the recognition of pathogens in dormantA. japonicus, thereby promoted the phagocytosis. The increased expression ofARPC5A,ARPC5B, andPSDpromoted phagocytosisviathe formation of vesicles for trafficking pathogen molecules to the phagosome, a crucial organelle for scavenging pathogens (Insallet al., 2001; Derrienet al., 2002;Trautweinet al., 2004; Watts, 2012). In addition, the upregulation of lysosomalGNSandGNSLaccelerated the hydrolysis of heparan sulfate, which is an important response to viral infection. Heparan sulfate was also found to be a crucial material in triggering autoimmunity. These alterations effectively limited pathogen infections (Wudunnet al., 1989; Freemanet al., 1992; Moket al., 2003; Barthet al., 2006; Piantaet al., 2017). Moreover, previous studies on sea cucumbers also showed a similar result, in which carbohydrate metabolism, lysozyme, and phagocytosis were upregulated during aestivation (Zhaoet al., 2014).Therefore, in the dormantA. japonicus, high levels of carbohydrate metabolism promoted phagocytosis, which is crucial for scavenging pathogens and improving survival during high temperatures.
Furthermore, the TPM value was used to characterize the pathway alterations (Trohaet al., 2018). In the dormantA. japonicus, high expressions ofPAMY,AMYB,AMY,andMGAMpromoted starch and sucrose hydrolysis to produce glucose, which is shunted into the TAC cycleviaglycolysis (Griffinet al., 1989; Marrelet al., 2002; Konget al., 2003; Simet al., 2008). An upregulation ofACADVLandACADLin the dormantA. japonicusdegraded longchain fatty acids to acetyl-CoA, an essential substrate in TCA cycle, for ATP synthesis (Ikedaet al., 1985; Izaiet al.,1992; Zhanget al., 2013; Shaoet al., 2015). A similar finding was observed in a previous study in which starch and fatty acids hydrolase were highly expressed during deep aestivation of sea cucumbers (Zhaoet al., 2014). These results suggest that the degradation of fatty acids and starch was the main route for providing energy in the dormant sea cucumbers. However, long-term consumption of stored fatty acid decreased the content of hexadecanoic acid,which further inhibited acetyl-CoA production and TCA cycle. The decreased levels of TCA cycle further confirmed that hypometabolism occurred in dormantA. japonicus.
In addition, PPI analysis predictedHK1interaction withVATPA, which accelerates H+into cell; however, they both enter the phagosome to improve phagocytic activities (Ciprianoet al., 2008). Thus, the high expression ofHK1indicates its function in improving the immune system of sea cucumbers during the aestivation period. Moreover, the phagocytosis-related genes, includingARPC5A,ARPC5B,PSD,GNS,GNSL, andMRC1, were upregulated in DG.This finding is consistent with previous results, in which the upregulation of carbohydrate hydrolase not only catalyzed starch degradation but also promoted phagocytosis.Optimal carbohydrate diets have significant stimulatory effects on phagocytosis (Pageet al., 1999; Argayosaet al.,2011). Therefore, our results indicate that the dormantA.japonicusconsumed carbohydrate and fatty acids mainly for energy supply. A high level of carbohydrate metabolism improved the expression of phagocytosis-related genes,which was beneficial for resisting pathogens during the aestivation. The results in this research were mainly from transcriptome analysis of the RNA level. In the future,detailed investigations are required to validate the findings of the present study.
5 Conclusions
In this study, transcriptome analysis was used to explore potential molecular mechanisms in dormantA. japonicus. Compared with gene expressions in RG, 2368 DEGs were identified in DG. Based on GO and KEGG analyses, the downregulated genes in DG were mainly annotated to DNA, RNA, and protein metabolic process. The PPI analysis further revealed that the downregulation of DNA, RNA, and protein metabolic processes reduced the motility, skeletal development, neural activity, cell proliferation, and development ofA. japonicus. In contrast, the upregulated genes were functionally belong to fatty acid and carbohydrate metabolisms. In the dormantA. japonicus, a high level of carbohydrate hydrolysis promoted phagocytosis, which is a primary innate immune response in invertebrates. Therefore, the improvement of phagocytosis facilitated theA. japonicusincreased resistance against pathogen invasion during the aestivation period. In addition, the decreases in oxoglutaric acid, acetyl-CoA, succinic acid, and citric acid indicated that hypometabolism occurred in the dormantA. japonicus. The findings of this study provide new insights into the mechanisms by whichA. japonicusare able to change their immune and physiological responses to acclimate to high temperatures.
Acknowledgements
This work was supported by the Science and Technology Program of Fujian Province (Nos. 2018R1003-1 and 2019R1013-5), the Special Funds for Marine and Fishery Structure Adjustment (No. 2020HYJG02), and the Marine Economy Innovation and Area Development Demonstration Project of Fujian Province (No. FJHJF-L-2020-4).
杂志排行

Journal of Ocean University of China的其它文章
- Evaluating the Accuracy of ERA5 Wave Reanalysis in the Water Around China
- Numerical Study of the Three Gorges Dam Influences on Chlorophyll-a in the Changjiang Estuary and the Adjacent East China Sea
- Parametric Stability Analysis of Marine Risers with Multiphase Internal Flows Considering Hydrate Phase Transitions
- Multi-Waves, Breathers, Periodic and Cross-Kink Solutions to the (2+1)-Dimensional Variable-Coefficient Caudrey-Dodd-Gibbon-Kotera-Sawada Equation
- Application of Improved Multi-Objective Ant Colony Optimization Algorithm in Ship Weather Routing
- Effect of Temperature on the Release of Transparent Exopolymer Particles (TEP) and Aggregation by Marine Diatoms(Thalassiosira weissflogii and Skeletonema marinoi)