不同培养条件下酿酒酵母菌的转录组差异分析
2021-03-06杨新陈莉杨双全卢红梅章之柱
杨新,陈莉,杨双全,卢红梅,章之柱
1(贵州大学,贵州省发酵工程与生物制药重点实验室,贵州 贵阳, 550025)2(贵州大学 酿酒与食品工程学院,贵州 贵阳, 550025)3(贵州大学 化学与化工学院,贵州 贵阳, 550025)4(开阳县市场监督管理局,贵州 贵阳, 550300)
硒(selenium,Se)是一种人体生长发育所必需的微量元素,具有“生命火种”、“心脏的守护神”和“抗癌之王”的美誉[1-2]。它是体内某些酶和蛋白的重要组成部分,具有抗衰老、抗氧化、抗肿瘤、影响人和动物的生殖发育、增强机体免疫力、拮抗有害重金属和预防多种疾病等生物学功能[1,3-5]。硒元素的缺乏会引起心血管疾病、肿瘤、克山病、大骨节病、癌症、高血压和免疫系统功能紊乱等疾病[6-9]。
酵母菌对许多微量元素具有较强的富集作用,比如铁、锌、硒、铬及锗等微量元素,但其富集机制与富集离子的种类有关[10-11]。此外,取决于酵母菌独特的细胞壁结构,它的主要结构及成分为葡聚糖、蛋白质、甘露聚糖、几丁质及少量脂类。因酵母菌具有体积小,表面积大,即比表面积大的特点,所以它具有代谢旺盛、繁殖速度快、产率高等优点。此外,还具有培养基原料来源广泛,易于人工控制培养,不受时间、季节、气候等条件的影响,且生产成本低、安全无污染、可持续性强等优点。因此,只需要在培养基中加入无机硒,酵母菌在生长繁殖过程中就可以将无机硒转化为有机硒,主要以蛋白质、氨基酸、多糖等结合而存在。故酵母菌是一种优越的富硒载体,富硒酵母菌作为一种理想的功能性食品添加剂,它不仅能提供硒源,而且还可以提供一定的蛋白质、氨基酸等营养物质。作为一种重要的、食品安全级微生物的酿酒酵母菌(Saccharomycescerevisiae)[12-13],将其作为富硒载体有着巨大的市场前景。
高通量测序技术又称“下一代”测序技术[14-15],随着该技术的发展,它在生物体转录组基因表达分析中被广泛应用,而且能够精确便捷地挖掘出相关功能基因[16-19]。转录组测序技术(RNA-sequencing,RNA-Seq)具有通量大、高分辨率、高灵敏度、不需克隆、检测范围广、成本低及操作简单等优点,因此成为转录组研究的主要手段[20-23],RNA-Seq技术已成功应用于多项研究中[24-26]。本实验采用转录组测序技术对酿酒酵母菌在不同硒浓度(0、20 μg/mL)培养条件下进行测序,然后基于转录组学采用生物信息学对酿酒酵母菌基因的表达差异进行分析,并对差异基因进行GO功能和KEGG富集分析,为今后对酿酒酵母菌富硒基因的挖掘以及研究提供了一定的理论基础。
1 材料与方法
1.1 材料与试剂
1.1.1 材料
具有富硒能力的酿酒酵母菌(实验室保藏)。
1.1.2 药品试剂
葡萄糖、硫酸镁,天津市永大化学试剂有限公司;酵母浸出粉、蛋白胨,上海博微生物科技有限公司;琼脂,北京Solarbio Science & Technology公司;亚硒酸钠,山东西亚化学股份有限公司;磷酸二氢钾,成都金山化学试剂有限公司;化学药品均为分析纯。
1.1.3 仪器与设备
SN-CJ-IF洁净工作台、YXQ-LS-5DS11立式压力蒸汽杀菌器,上海博讯实业有限公司医疗设备厂;SPX-250B智能型生化培养箱、DHG—9140B(101-2B)智能型电热恒温鼓风干燥箱,上海琅玕实验设备有限公司;TG16-WS台式高速离心机,湖南湘仪实验室仪器开发有限公司;BCD-290 W冰箱,青岛海尔股份有限公司;SZ-96A自动纯水蒸馏器,上海嘉措仪器设备有限公司;ZD-2A自动电位滴定仪,上海大普仪器有限公司;ESJ220-4B电子天平,沈阳龙腾电子有限公司。
1.2 实验方法
1.2.1 培养基配制
YPD培养基(g/L):葡萄糖10.0,蛋白胨20.0,酵母浸出粉10.0,pH自然。
基础发酵培养基(g/L):葡萄糖20.0、酵母浸出粉10.0、蛋白胨20.0、KH2PO4·3H2O 3.0、MgSO4·7H2O 1.0,121 ℃条件下高压灭菌20 min,pH 4.5,备用。
1.2.2 菌体收集
将酿酒酵母菌种子液以6%的接种量分别接种于硒质量浓度为0、20 μg/mL(每个硒浓度做3个重复)的富硒培养基中,培养基初始pH值为4.5,加硒时间为培养后6 h,装液量为100 mL/250 mL锥形瓶,在30 ℃,150 r/min的恒温摇床上培养72 h。培养结束后,离心收集菌体,分别标记为Kb1、Kb2、Kb3和Se1、Se2、Se3,然后立即置于液氮中保存。
1.2.3 酵母菌总RNA提取与质量检测
根据制造商的说明,使用TRIzol®试剂盒从酿酒酵母菌细胞中提取总RNA,并使用DNase I去除基因组DNA。对提取得到的RNA样品,使用1%(质量分数)的琼脂糖凝胶电泳检测RNA的纯度和完整度,使用NanoDrop2000 RNA检测RNA的纯度及浓度,使用Agilent 2100 Nano检测RNA的完整值(RNA integrity number,RIN)。仅使用高质量的RNA样品来构建测序文库,然后上机测序。
1.2.4 文库制备和转录组测序
RNA纯化,反转录,文库构建和测序均根据制造商的指导在上海美吉生物医药科技有限公司进行。使用Illumina TruSeqTM RNA Sample Prep Kit试剂盒构建RNA-seq转录组文库。立即根据poly(A)选择方法,通过oligo-dT磁珠分离mRNA。首先通过片段缓冲液进行片段化,然后使用SuperScript double-stranded cDNA合成试剂盒和随机六聚体引物合成双链cDNA。再根据文库构建方案对合成的cDNA进行末端修复,即磷酸化和“A”碱基添加,选择大小为2%低范围超琼脂糖上200~300 bp的cDNA目标片段,然后使用Phusion DNA聚合酶进行15个PCR循环进行PCR扩增。通过TBS380定量后,用Illumina Novaseq 6000(2×150 bp读长)对双端RNA-seq测序文库进行上机测序。
1.3 数据分析
使用软件SeqPrep(https://github.com/jstjohn/SeqPrep)和Sickle(https://github.com/najosh-i/sickle)以默认参数对原始的末端读数进行裁剪和质量控制。使用软件TopHat(http://tophat.cbcb.umd.edu/,版本2.1.1)[27]以定向模式将干净的读数分别与参考基因组比对。基于所选参考基因组序列,使用StringTie(http://ccb.jhu.edu/software/stringtie/)软件对Mapped Reads进行拼接,然后与原有的基因组注释信息进行比较。使用DESeq2软件对Raw counts进行统计分析,基于P<0.05 & |log2FC|≥2为筛选条件,筛选获得实验组与空白组之间表达差异的基因。
聚类分析是根据不同条件下差异基因TPM值的表达水平,作层次聚类分析,用于判断DEGs在不同条件下的表达模式。将筛选获得的差异表达基因在GO数据库和KEGG数据库中进行比对,然后根据比对结果进一步分析基因产物的功能及在细胞中的代谢途径。
2 结果与分析
2.1 RNA样品质量分析
转录组测序的RNA样品质量要求OD260/OD280值应为1.8~2.2、OD260/OD230值>2时核酸的纯度较高,RIN为1~10,数值越接近10完整性越好[28]。由图1及表1可知,6个样品RNA的纯度、浓度和完整度等质量指标完全符合后续转录组测序的要求,因此可以构建cDNA文库,进行上机测序。
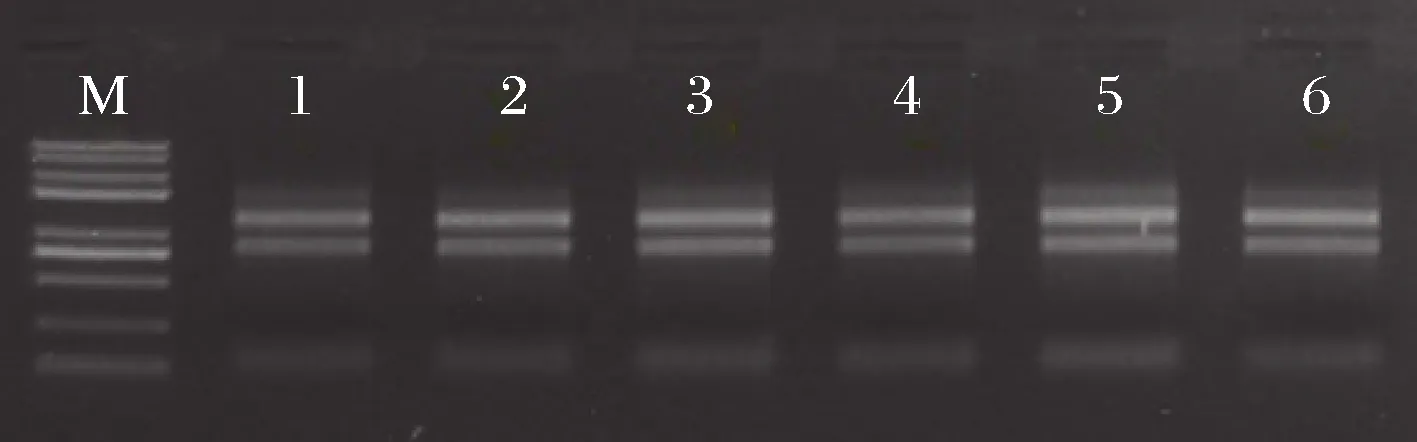
M-marker;1~6-样品Kb1、Kb2、Kb3、Se1、Se2、Se3图1 富硒酵母RNA样品琼脂糖凝胶电泳图Fig.1 Agarose gel electrophoretogram of Se-enriched yeast RNA samples

表1 酿酒酵母RNA样品质量检测结果Table 1 Quality test results of RNA samples of saccharomyces cerevisiae
2.2 测序数据质量评估与分析
为探究酿酒酵母菌在不同硒浓度培养条件下的变化机制,首先对其进行转录组测序,共获得261 794 672条原始序列,然后对原始序列进行过滤处理,共获得259 164 884条干净序列。由表2可知,Kb组和Se组的Q20平均值分别为98.90%和98.89%,Q30平均值分别为96.15%和96.12%,此外GC相对含量分别为42.48%和42.23%。因此可知,转录组测序数据质量好,准确性高,可用于后续分析。
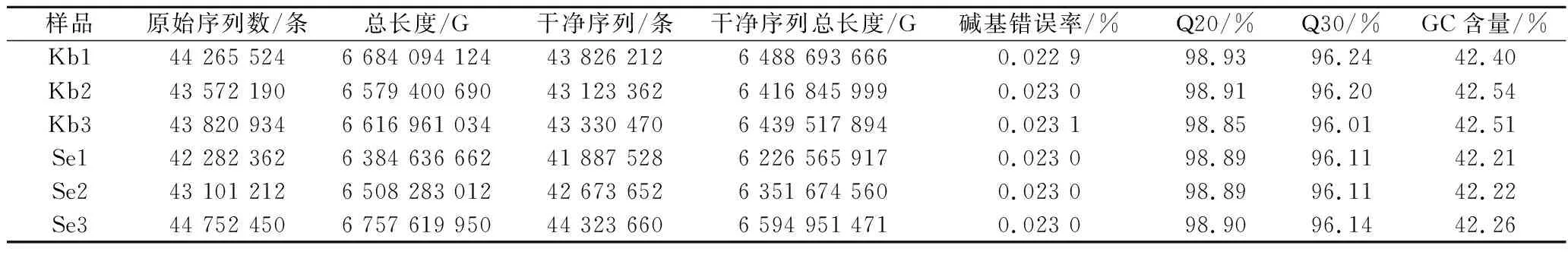
表2 测序数据统计表Table 2 Statistical table of sequencing data
2.3 参考序列比对分析
2.3.1 比对结果统计
本研究选取NCBI中酿酒酵母菌的基因组作为参考基因组。通常情况下,如果参考基因组选择合适,而且相关实验不存在污染,实验所产生的测序干净序列能定位到基因组上的比率通常会高于65%。本研究用Smalt软件对高质量数据进行比对,其结果见表3。
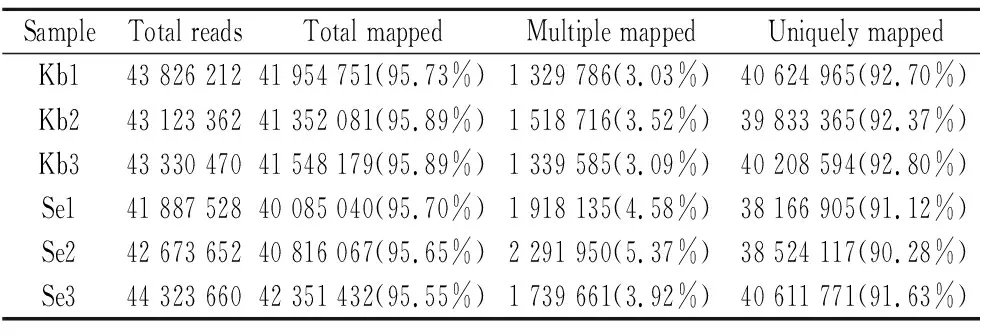
表3 比对结果统计表Table 3 Statistical table of comparison results
由表3可知,Kb组和Se组转录组测序获得的干净序列中,能定位到基因组上的干净序列片段平均值分别为41 084 180条和41 618 337条,占比为95.55%~95.89%,在参考序列上有多个比对位置的干净序列片段平均值分别为1 396 029条和1 983 249条,占比为3.03%~5.37%,在参考序列上有唯一比对位置的干净序列片段平均值分别为40 222 308条和39 100 931 条,占比为90.28%~92.8%。其中能定位到基因组上的干净序列片段数均大于95%,而在参考序列上有多个比对位置的干净序列片段数均小于10%,因此本次研究所测序列没有受到污染。
2.3.2 测序饱和度分析
转录本的表达水平不同,准确定量所需的测序深度也有所区别,在低表达的转录本中,为了保证定量的准确性,往往需要更大的测序深度,因而,可以通过饱和度曲线来评估不同测序深度条件下不同表达水平转录本是否被准确定量。本研究采用RSeQC-2.3.6软件对6个样品的测序饱和度进行分析,其结果如图2所示。
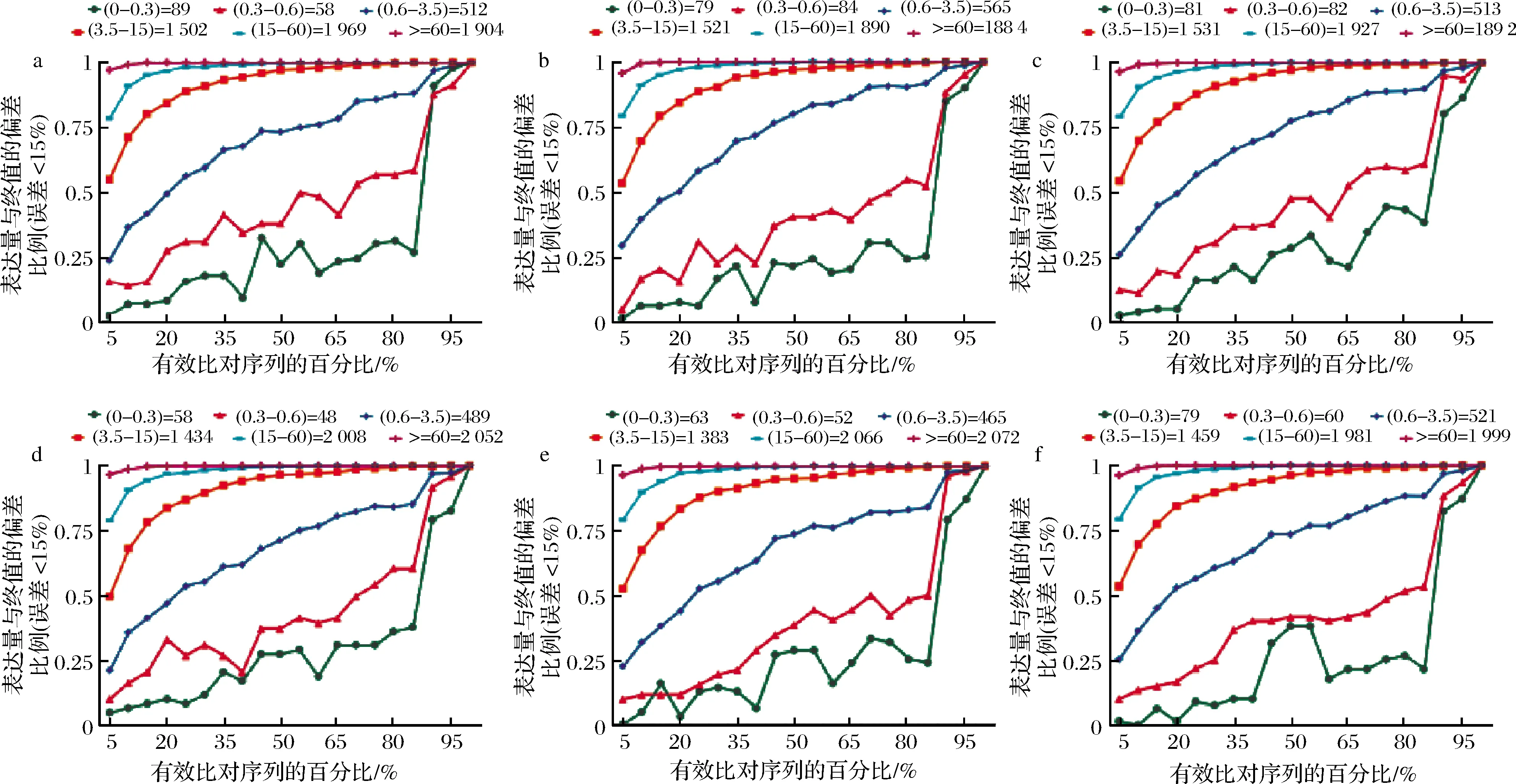
a-Kb1饱和度;b-Kb2饱和度;c-Kb3饱和度;d-Se1饱和度;e-Se2饱和度;f-Se3饱和度图2 测序饱和度曲线图Fig.2 The curve map of sequencing saturation
通常情况下,当TPM值>3.5的基因,在测序序列的40%比对上时接近饱和,即纵轴数值趋近于1,说明饱和度总体质量较高,且转录本表达水平越高相对误差越小。由图2可知,本研究中所有测序量能够覆盖绝大多数的表达基因,因此,该测序深度条件下转录本能够被准确定量。
2.3.3 测序覆盖度分析
测序覆盖度是用来评估测序结果的均一性,它是针对样品中所有基因的5′~3′区域上序列覆盖情况的综合分析。本研究采用RSeQC-2.3.6软件对本研究的6个样品进行分析,其结果如图3所示。其中横坐标为单个基因的碱基长度占总碱基长度的百分比,纵坐标为比对到所有基因的横轴位置上相应区间内的序列条数的总和。由图3可知,该测序所得序列在基因上均匀分布,且测序无偏向性。
图3 测序覆盖度分布图Fig.3 The distribution map of sequencing coverage
2.3.4 不同区域序列分布
将比对到基因组上的序列在参考基因组不同区域的分布情况进行统计,主要定位区域为5个,即编码区(coding sequence,CDS)、内含子(intron)、基因间区(intergenic region)和5′和3′非翻译区(untranslated region,UTR)。如果测序序列被定位到内含子上,通常情况是因为注释不完全的基因组或是有非成熟的mRNA污染。如果测序序列被定位到基因间隔区域时,通常是因为背景噪音或者注释不完全的基因组。
图4分别为每个样品的序列在参考基因组不同区域的分布情况,可以看出6个样品的序列定位到编码区域最多,分别为98.27%、97.82%、98.23%、97.94%、96.93%和98.80%;定位到内含子区域分别为0.13%、0.13%、0.15%、0.15%、0.15%和0.15%;定位到基因间隔区域分别为0.05%、0.05%、0.05%、0.06%、0.06%和0.06%;定位到5′和3′非翻译区域分别为1.55%、2.01%、1.58%、1.85%、2.87%和0.98%。
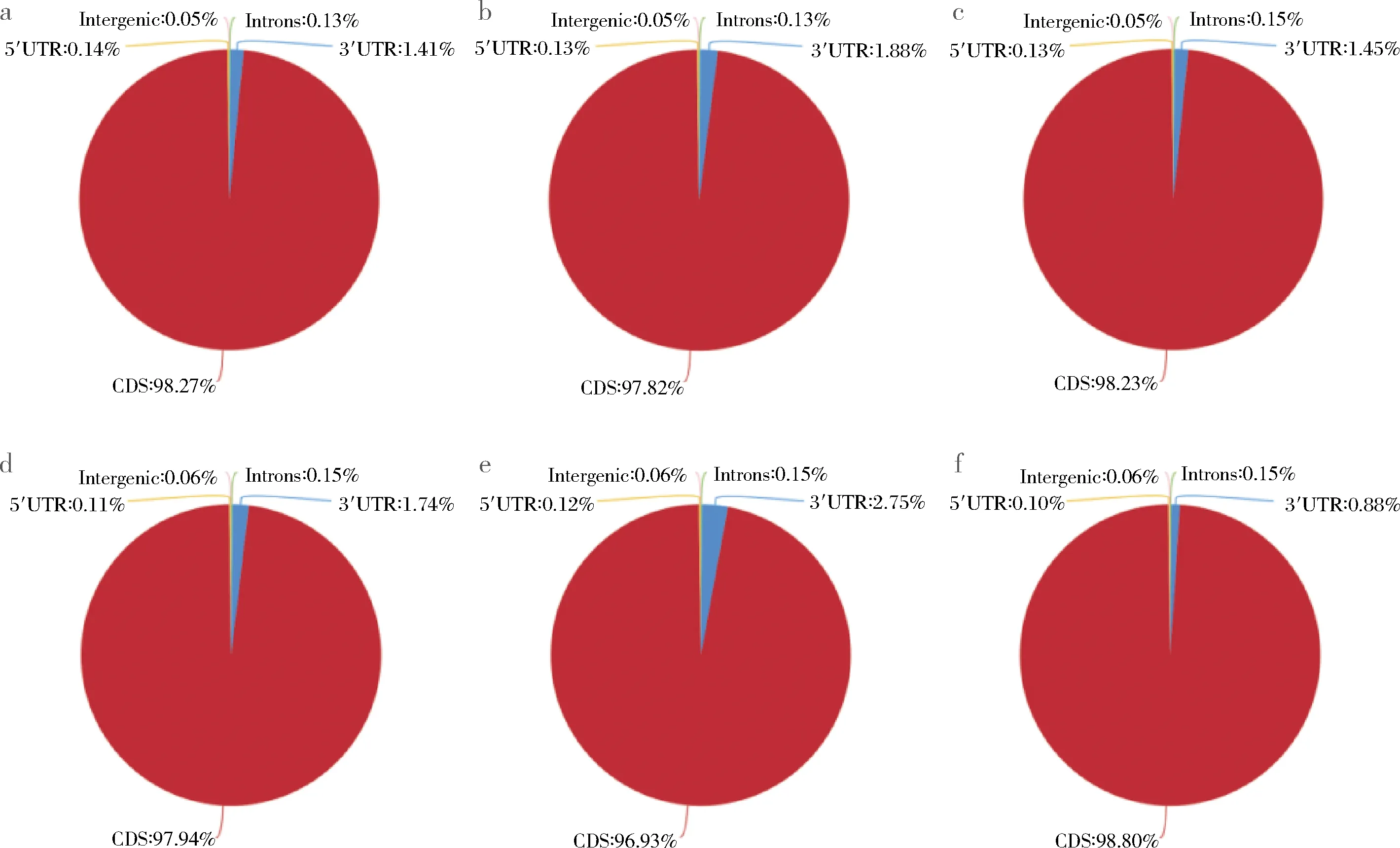
a-Kb1;b-Kb2;c-Kb3;d-Se1;e-Se2;f-Se3图4 不同区域Reads分布统计饼图Fig.4 Statistical pie charts of Reads distribution in different regions
2.3.5 不同染色体序列分布
为了从宏观上了解所测序列在各染色体上的分布情况,因此将比对到基因组上的序列在参考基因组不同染色体上的分布情况进行统计。图5为6个样品比对到基因组上的序列在不同染色体上的分布统计柱状图,其中横坐标为不同染色体的名称,纵坐标为序列在染色体上的数量。由图5可知,酿酒酵母菌富硒后,NC-001136.10号染色体的序列数变化较大,而其他染色体的变化较小。
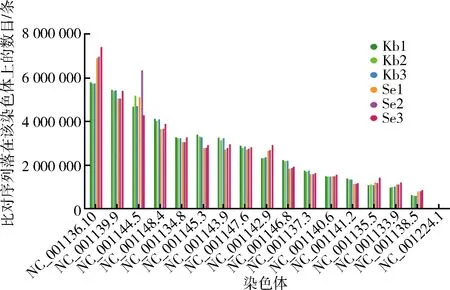
图5 不同染色体序列分布统计柱状图Fig.5 Statistical histogram of reads distribution on different chromosomes
2.4 转录组功能注释
将酿酒酵母菌组装转录本分别在GO、KEGG、COG、NR、Swiss-Prot和Pfam各大数据库的比对结果进行统计(见表4)。其中有1 401个(21.74%)Unigene被注释到GO数据库,有3 665个(56.87%)Unigene被注释到KEGG数据库,有5 630个(87.35%)Unigene被注释到COG数据库,有6 112个(94.83%)Unigene被注释到NR数据库,有6 077个(94.29%)Unigene被注释到Swiss-Prot数据库和有5 059个(78.49%)Unigene被注释到Pfam数据库。
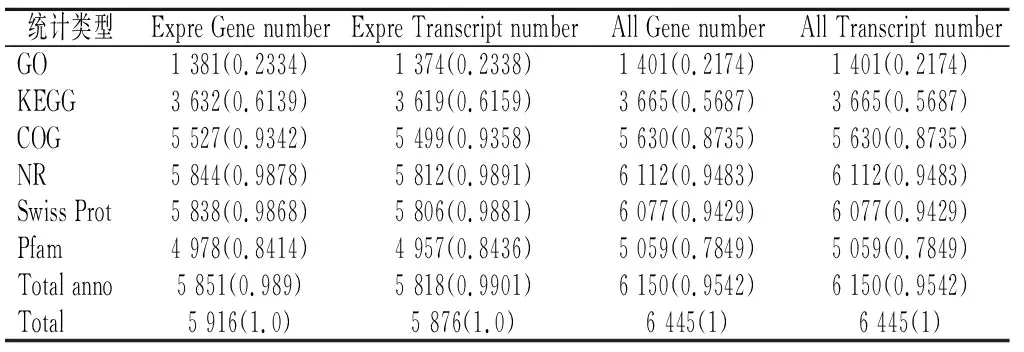
表4 功能注释统计表 单位:%Table 4 Functional annotation statistics table
2.5 样本间相关性分析
在研究过程中,样品之间基因表达水平相关性通常用皮尔逊相关系数的平方(R2)来表示,相关系数R2的值为0~1,越接近1,表明样品之间的相关性越大,即相似度越高。由图6可知,本研究中6个样品之间的相关系数R2的最小值为0.943,故本研究中结果可靠,同时对样品的选择也合理。
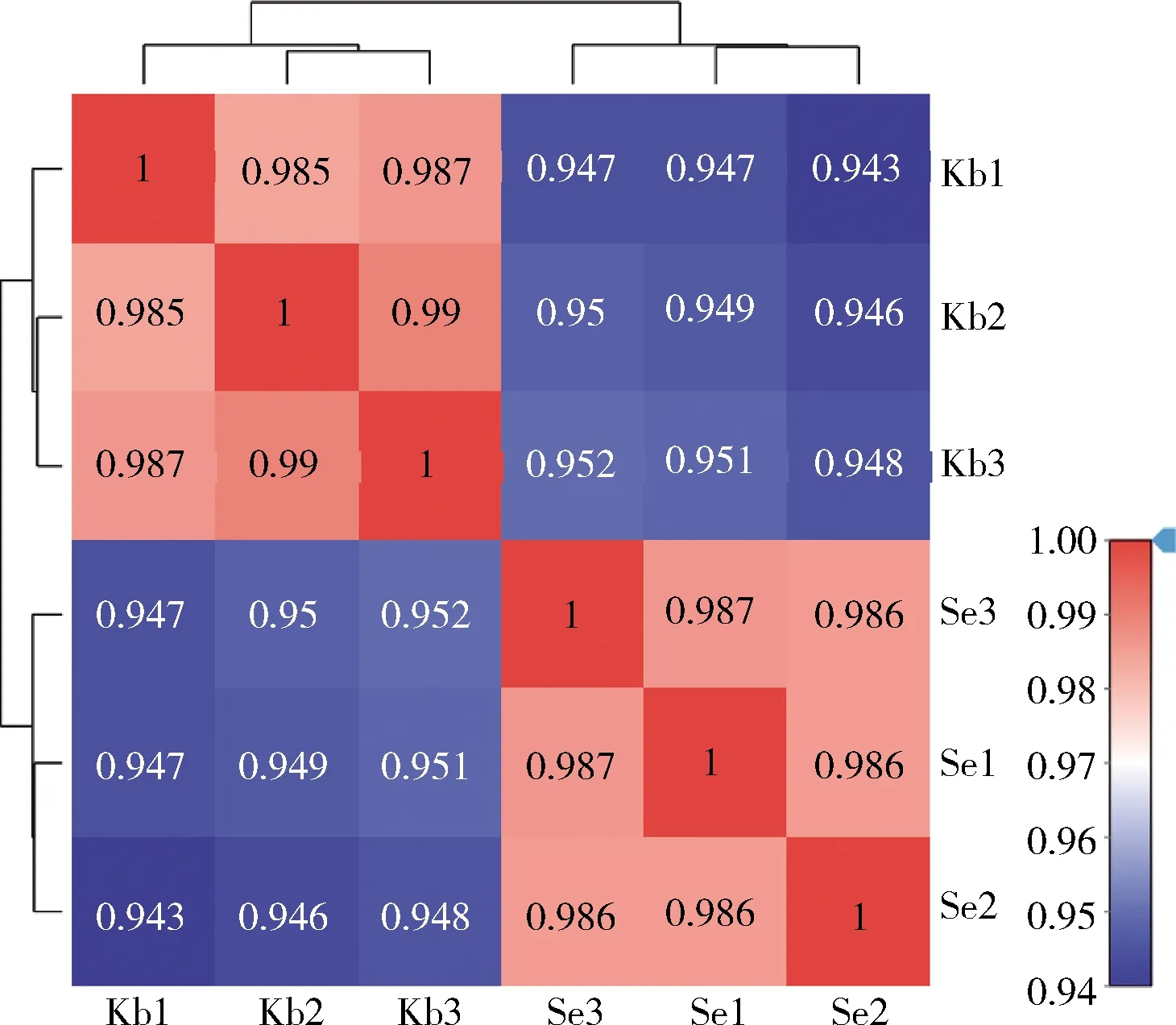
图6 样品间相关性热图Fig.6 Heat map of correlation between samples
2.6 基因表达水平分析
2.6.1 差异表达基因筛选
同一生物体在不同时间和环境条件下,其某些基因的表达会存在显著性差异。同样,酿酒酵母菌从无硒环境到有硒环境中,主要通过调节机体内的某些基因的表达,来适应有硒环境。因本研究中有生物学重复的样品,其基因差异分析应使用Bioconductor软件包的DESeq2(V1.6.3)进行分析[29-30],设置P≤0.05,上/下调差异倍数为≥2.0倍。共筛选出差异表达的基因994个,其中有498个基因表现为下调,占总差异表达50.10%,有496个基因表现为上调,占总差异表达49.90%,其结果分析见差异基因火山图及散点图(图7)。

a-火山图;b-散点图图7 表达量差异火山图及散点图Fig.7 Expression difference volcanic map and scatter map
通过对基因在各样品中的表达进行聚类,通常表达模式相似的基因具有功能相关性,因此,可以根据聚类分析来判断差异基因在不同实验条件下的表达模式,从而推断未知基因的功能。本研究采用差异分析软件DESeq2进行分析,以P≤0.05,上/下调差异倍数为≥10.0倍进行筛选,得到55个差异显著基因,然后绘制基因热图,结果如图8所示。

图8 基因聚类热图Fig.8 Heat map of gene cluster
2.6.2 差异基因GO富集分析
对筛选出来的差异表达基因进行GO富集,可以查看其参与的生物学过程、构成细胞的组分及分子功能等信息,因此,对差异表达基因进行GO功能显著性富集分析,可以得到差异表达基因与哪些生物学功能显著相关。
酿酒酵母菌在不同硒质量浓度(0、20 μg/mL)培养条件下的差异基因GO富集分类结果如图9所示,结果显示共分为41个功能组,其中,生物过程(biological process,BP)有17个功能亚类,主要注释为细胞过程、代谢过程、单一生物过程、细胞成分的组织或生物发生等亚类;细胞组成有13个功能亚类,主要注释为细胞、细胞部分、细胞器、细胞器部分、大分子复合物等亚类的组成;分子功能有11个功能亚类,主要注释为催化活性、捆绑等亚类。

图9 GO分类统计柱形图Fig.9 Statistical histogram of GO classification
2.6.3 差异基因KEGG富集分析
生物体内的生物学功能主要通过不同基因的相互协调而发挥作用,通过对差异表达基因进行通路显著性分析,可以找出差异表达基因相对于所有有注释的基因显著富集的通路,同时还可以确定差异表达基因参与的最主要生化代谢途径和信号转导途径。
酿酒酵母菌在不同硒浓度(0、20 μg/mL)培养条件下的差异基因KEGG富集气泡图是KEGG富集分析结果的可视化方式。纵坐标表示通路名称,横坐标表示比值,其值越大,该代谢通路富集的程度越大,而气泡的大小表示此通路中基因数量,气泡的颜色对应于不同的q值范围。本研究挑选最显著的20条富集基因进行展示。由图10可知,富集到“减数分裂-酵母”通路上的基因共有38个,呈显著富集;其次是“糖酵解/糖异生”、“乙醛酸和二羧酸酯代谢”、“过氧化物酶体”、“淀粉和蔗糖代谢”与“氨基糖和核苷酸糖代谢”通路上的基因分别有24、19、19、17、16个;富集到“半乳糖代谢”与“丙酮酸代谢”通路上的基因均为14个;富集到“甲烷代谢”通路上的基因有12个;富集到“脂肪酸降解”通路上的基因有11个;富集到“果糖和甘露糖代谢”通路上的基因有10个;富集到“谷胱甘肽代谢”与“甘油脂代谢”通路上的基因均有9个;富集到“脂肪酸生物合成”与“丙酸酯代谢”通路上的基因均为7个;富集到“缬氨酸,亮氨酸和异亮氨酸的降解”与“β-丙氨酸代谢”通路上的基因均为6个;富集到“不饱和脂肪酸的生物合成”与“戊糖和葡萄糖醛酸酯的相互转化”通路上的基因均为5个;富集到“抗坏血酸和藻酸盐代谢”通路上的基因为3个。

图10 KEGG富集分析气泡图Fig.10 Bubble map of KEGG enrichment analysis
3 结论
本研究采用Illumina HiSeq测序平台,对酿酒酵母菌在不同硒质量浓度(0、20 μg/mL)培养条件下的转录组进行测序分析,测序质量良好。其中对照组是以不富硒的酿酒酵母菌为材料,共获得131 658 648条原始序列,经过过滤后得到130 280 044条干净序列;实验组是以富硒的酿酒酵母菌为材料,共获得130 136 024条原始序列,经过过滤后得到128 884 840 条干净序列。基于P<0.05 & |log2FC|≥2为筛选条件,2组样品之间共筛选获得994个显著性差异表达基因,其中有496个基因表达量上调,有498个基因表达量下调。通过差异基因GO富集分析结果可以明确酿酒酵母菌在不同硒浓度培养条件下的细胞组分、生物学过程和分子功能三大分类中的差异表达基因。最后对差异表达基因进行KEGG富集分析,确定了差异表达基因的显著富集通路,同时找出了差异表达基因参与的最主要生化代谢途径和信号转导途径,最终以气泡图的形式对分析结果进行展示。结果表明2组样品的差异表达基因KEGG代谢通路中显著富集的通路有:减数分裂-酵母、糖酵解/糖异生、乙醛酸和二羧酸酯代谢、过氧化物酶体、淀粉和蔗糖代谢、氨基糖和核苷酸糖代谢、硫代谢、半胱氨酸和蛋氨酸代谢、谷胱甘肽代谢及硒化合物代谢等。研究结果为进一步分析酿酒酵母菌富硒基因的挖掘和研究提供了一定理论参考及科学依据。
