Dynamic evolution of transposable elements,demographic history,and gene content of paleognathous birds
2021-03-01ZongJiWangGuangJiChenGuoJieZhangQiZhou
Zong-Ji Wang ,Guang-Ji Chen ,Guo-Jie Zhang,5,6,7,* ,Qi Zhou
1 Institute of Animal Sex and Development, Zhejiang Wanli University, Ningbo, Zhejiang 315100, China
2 MOE Laboratory of Biosystems Homeostasis & Protection, Life Sciences Institute, Zhejiang University, Hangzhou, Zhejiang 310058,China
3 Department of Neuroscience and Developmental Biology, University of Vienna, Vienna 1090, Austria
4 BGI-Shenzhen, Beishan Industrial Zone, Shenzhen, Guangdong 518083, China
5 State Key Laboratory of Genetic Resources and Evolution, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming,Yunnan 650223, China
6 Section for Ecology and Evolution, Department of Biology, University of Copenhagen, Copenhagen DK-2100, Denmark
7 Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming, Yunnan 650223, China
8 Center for Reproductive Medicine, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310058,China
ABSTRACT Palaeognathae includes ratite and tinamou species that are important for understanding early avian evolution.Here,we analyzed the whole-genome sequences of 15 paleognathous species to infer their demographic histories,which are presently unknown.We found that most species showed a reduction of population size since the beginning of the last glacial period,except for those species distributed in Australasia and in the far south of South America.Different degrees of contraction and expansion of transposable elements (TE) have shaped the paleognathous genome architecture,with a higher transposon removal rate in tinamous than in ratites.One repeat family, AviRTE, likely underwent horizontal transfer from tropical parasites to the ancestor of little and undulated tinamous about 30 million years ago.Our analysis of gene families identified rapid turnover of immune and reproductionrelated genes but found no evidence of gene family changes underlying the convergent evolution of flightlessness among ratites.We also found that mitochondrial genes have experienced a faster evolutionary rate in tinamous than in ratites,with the former also showing more degenerated W chromosomes.This result can be explained by the Hill-Robertson interference affecting genetically linked W chromosomes and mitochondria.Overall,we reconstructed the evolutionary history of the Palaeognathae populations,genes,and TEs.Our findings of co-evolution between mitochondria and W chromosomes highlight the key difference in genome evolution between species with ZW sex chromosomes and those with XY sex chromosomes.
Keywords:Paleognaths; Demographic history;Transposable elements;Gene families;Mitochondria
INTRODUCTION
Modern birds include Palaeognathae and Neognathae,which diverged from each other over 110 million years ago (Jarvis et al.,2014),with the former currently comprising only 5.7% of extant bird species.Palaeognathae includes the flightless ratites and volant tinamous.Ratites are broadly distributed across the major continents,and includeApterygidae(kiwis)and four families of species with much larger body size than any other bird species:i.e.,Struthionidae(ostriches),Rheidae(rheas),Casuariidae(cassowaries),andDromaiidae(emus)(Angst & Buffetaut,2017).As well as in Africa,ostriches also used to be present in a large part of Eurasia (Europe and Asia) (Houde,1986).Emus,cassowaries,and kiwis are native to Australasia and rheas are distributed in South America.Tinamous consist of 46 extant species in nine genera,which are exclusively located in Central and South America(Figure 1).Despite the evolutionary success of these birds in terms of species diversity and geographic range,their demographic histories remain largely unknown.
Paleognathous species are important for understanding the early evolution of birds due to their unusual features of gigantism and flightlessness,resulting in a lower genome-wide evolutionary rate than that of other birds (Wang et al.,2019).For example, cytogenetic studies and recent genome analyses of ostriches found fewer intragenomic rearrangements compared to that in other birds (O’Connor et al.,2018; Takagi et al.,1972).The loss of flight capability and the increase in body size (gigantism) seem to have occurred independently multiple times among ratites (Sackton et al.,2019).However,the genomic bases for these changes remain poorly understood,except for several recent studies on emus and rheas (Sackton et al.,2019; Young et al.,2019).Tinamous do not exhibit powered flight,but are capable of short,burst flapping flight,which may be due to their smallest heart-to-body size ratio among birds (Altimiras et al.,2017;Bishop,1997).It has been proposed that the metabolic requirement for powered flight is linked to a reduced genome size (Altimiras et al.,2017; Kapusta et al.,2017; Wright et al.,2014),as evidenced by the smaller genomes of bats and birds relative to those of other mammals and birds.In addition to the different rates of genomic deletions between the two groups of species, the contribution of transposable element (TE)turnover to the variable genome sizes of paleognaths remains to be elucidated,as previous analysis included only ostrich and white-throated tinamou (Kapusta et al.,2017).Whether gene content diversity among species has contributed to the convergent evolution of flightlessness among ratites also remains to be clarified.
Paleognaths provide a unique model for studying the evolutionary history of bird sex chromosomes.Of note,most paleognathous species have homomorphic sex chromosomes with cytologically indistinguishable Z and W chromosomes(Ogawa et al.,1998; Pigozzi,1999; Pigozzi & Solari,1999;Shetty et al.,1999; Tsuda et al.,2007),in contrast to the highly heteromorphic sex chromosomes of Neognathae and mammals. The reason for this unusually slow sex chromosome differentiation is unclear.An important distinction between the ZW and XY sex system is that mitochondria,which are non-recombining and are specifically transmitted through females,are genetically linked to the W chromosome(Berlin et al.,2007).The Hill-Robertson interference,i.e.,interference between linked loci reducing the efficacy of selection,is therefore expected to act on both mitochondrial DNA (mtDNA) and W chromosomes (Charlesworth &Charlesworth,2000).However,few genomic studies have examined whether the extent of sex chromosome differentiation is correlated with the mitochondrial gene evolutionary rate,except for an earlier study on collared flycatchers (Smeds et al.,2015).
In this study,we analyzed the whole-genome sequences of 15 paleognathous female birds to reconstruct the evolutionary history of their populations,TEs,and genes.We characterized dynamic expansions and contractions of TE and gene families in these birds.We further demonstrated that the evolutionary rates of mitochondria and W chromosomes were highly correlated.
MATERIALS AND METHODS
Data description
Assembled sequences (both nuclear and mitochondrial genomes) and gene annotations of 15 species of tinamous and ratites (sample information:Supplementary Table S1)were retrieved from our previous study (Wang et al.,2019).All analyses performed were based on our recently produced phylogenomic tree using whole-genome non-coding sequence alignments (Wang et al.,2019).
Here, we presented the genomic annotations of 12 Palaeognathae species (Supplementary Table S1),together with previously reported genomes (Le Duc et al.,2015; Wang et al.,2019; Zhang et al.,2014),representing all extant Palaeognathae orders (Handford & Mares,1985).Samples were selected to cover the Tinamiformes order,with nine species within four genera represented for the first time(Figure 1).We produced the single nucleotide polymorphism(SNP) sites for each studied species,which were used to reconstruct their demographic history (see below for detailed methods).
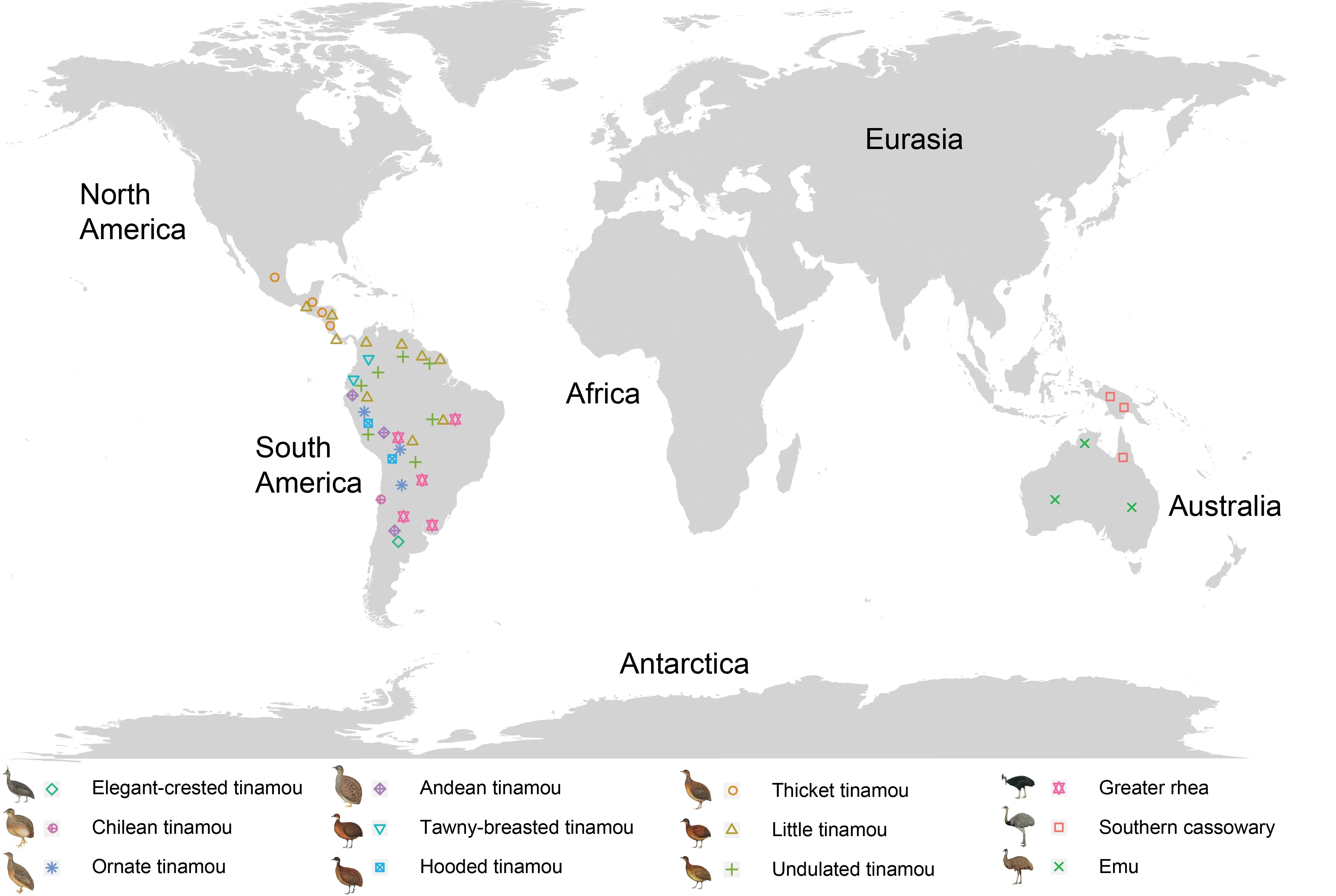
Figure 1 Continental distributions of paleognathous species investigated in this study
Demographic history analysis
We investigated the demographic histories of the different paleognathous species with whole-genome datasets using the pairwise sequentially Markovian coalescent (PSMC) approach(Li & Durbin,2011),which can infer changes in effective population size (Ne) within 10 000 to 1 000 000 years.We performed PSMC (Li & Durbin,2011) with the heterozygous SNP loci produced by the GATK pipeline (DePristo et al.,2011). Parameters were set to “N30–t5–r5–p 4+30*2+4+6+10” and bootstrapping (100 times) was performed for each species to determine variance inNeestimates.We used the estimated values of generation time and mutation rate to scale the results.We applied branchspecific estimates of the synonymous substitution rate per synonymous site (dS) from our dated phylogeny as proxies for the rate of mutation.The age of sexual maturity (collected from published results) was multiplied by a factor of two as a proxy for generation time (Handford & Mares, 1985)(Supplementary Table S2).
Evolutionary analyses of transposable elements
We used RepeatMasker (v.4.0.7) with RepBase (20 160 829)and thede novoprediction program RepeatModeler (v.open-463 1.0.8) to predict and categorize repetitive elements.We used the Kimura 2-parameter distance to estimate the divergence level between individual TE copies and their consensus sequences.Kimura distances between genome copies and TE consensus were calculated using the RepeatMasker (v.4.0.7) built-in scripts(calcdivergencefromalign.pl).We dated the expansion of certain repeat subfamilies based on their sequence divergence patterns in the phylogeny by parsimony.We inferred more recent or ancestral subfamilies if they showed a sequence divergence pattern concentrated at a lower or higher divergence level and shared among species or specific to certain lineages.To test the influence of assembly quality on the estimation of TE abundances,we performed TE annotation with the same pipeline on a newly produced emu genome using PacBio and Hi-C technologies.We first annotated the genomic copies of AviRTE (a novel family of long interspersed elements) for each Palaeognathae genome assembly using RepeatMasker (v.open-4.0.6) based on its published consensus sequences (Suh et al.,2016).We then used all of the predicted AviRTE sequences from all studied paleognathous genomes to reconstruct the putative ancestral consensus sequence by RepeatModeler (v.open-1.0.8) to calculate the divergence of AviRTE from each species.To date the time of horizontal transfer,we compared the flanking sequences (a block of Ns with the same length) of each AviRTE copy between species by blat (version 36) (Kent,2002) using those of AviRTEs fromCrypturellus souiwith the highest number of copies as queries.We defined a pair of orthologous AviRTEs if their flanking sequences were aligned to each other.
Evolutionary analyses of gene families
To examine the evolution of gene families in paleognathous birds,genes from ratites,tinamous (15 species in total),and chicken (Ensembl release-87) were clustered into gene families by Treefam (Li et al., 2006) (min_weight=10,min_density=0.34,and max_size=500).Family expansion or contraction analysis was performed by CAFÉ (De Bie et al.,2006).Gene Ontology (GO) enrichment analysis was carried out based on the algorithm implemented in GOstat (Beißbarth& Speed,2004).The computedP-value was then adjusted for multiple tests by specifying a false discovery rate (<0.05)using the Benjamini-Hochberg method (Benjamini et al.,2001).
Evolutionary and association analyses of mitochondrial genes
The mitochondrial protein-coding genes were identified using MITOS (Bernt et al.,2013) and curated by comparison with known sequences of other published ratites and tinamous from GenBank.For the PAML analyses,we first assigned orthologous relationships among all paleognathous birds and the outgroup (chicken) using the reciprocal best blast hit algorithm and syntenic information. We used PRANK(Löytynoja & Goldman,2005) to align the orthologous gene sequences,which takes phylogenetic information into account when placing a gap into the alignment.We filtered the PRANK alignments by gblocks (Talavera & Castresana,2007) and excluded genes with a high proportion of low complexity or repetitive sequences to avoid alignment errors.We estimated the evolutionary rates of all individual mitochondrial datasets for each branch using the Codeml program with the free-ratio model in the PAML package (Yang,2007).We tested the association between the evolutionary rate of mitochondrial genes and sex chromosome differentiation degree using the phylogenetic generalized least squares (PGLS) model implemented in R package APE (Paradis et al.,2004) and nlme (https://CRAN.R-project.org/package=nlme).
RESULTS
Temporal changes in paleognathous species populations
We found large variation inNeamong species,as indicated by their different mean values over time (Supplementary Table S3),which ranged from 20 000–40 000 in emus,Southern cassowaries,and ornate tinamous to approximately 160 000–1 800 000 in elegant-crested and white-throated tinamous.Similarly,the minimumNeestimated over the time span analyzed varied between 5 000–9 000 (emus and Southern cassowaries) to >50 000 (elegant-crested and white-throated tinamous), and the maximumNevaried between 30 000–60 000 (e.g., emus and Andean tinamous) to 700 000–750 000 (undulated tinamous).Similar to the results reported for neognaths (Nadachowska-Brzyska et al.,2015),most species showed a sharp decline in population size since the beginning of last glacial period (LGP) about 100 000 years ago (Figure 2),except for species in the far south of the Southern Hemisphere and Australasia.
Two species (brown kiwis and hooded tinamous) in our study are classified as vulnerable on the IUCN Red List of Threatened Species (http://www.iucnredlist.org/),and another two species (white-throated tinamous and greater rheas) are classified as near threatened (Supplementary Table S3).Their PSMC patterns exhibited a different trend to their historical population changes (Figure 2).Brown kiwis showed a stableNeduring the LGP.Hooded tinamous had a relatively highNe(~140 000) but experienced a continuous reduction inNe30 000 years ago.The white-throated tinamous,which are classified as near threatened,experienced long-term decrease inNefrom 270 000 to 50 000.Greater rheas showed a rather stableNefor a long time until 40 000 years ago,when they experienced a drastic reduction inNeto 50 000.Our results suggest that these vulnerable and near threatened species experienced a population size reduction at the end of the LGP,predating the recent declines impacted by human activities.
Temporal evolution of TEs in paleognaths
With similar sequencing coverage,the assembled genome sizes of the ratites were, on average, 27.72% larger(1.276±0.177 vs.1.005±0.035 Gb,P=0.001 2,Wilcoxon test)than those of the flighted tinamous (Supplementary Table S1).Comparison of emu genomes produced by Illumina and PacBio sequences indicated that the annotated repeat sequences, except for the long terminal repeat (LTR)elements,were similar in size,suggesting that most TEs in the studied Palaeognathae genomes were properly annotated(Supplementary Table S4).Repeat library-based annotations of genome samples showed low variance in genomic repeat content and a relatively constant diversity of repeat types.We found that paleognaths generally had a similar level of genome-wide repeats,ranging from 4.89% in thicket tinamous to 5.54% in ostriches,which was largely attributed to their similar long interspersed nuclear element (LINE) content(Supplementary Table S5).Ratites had significantly (P<0.000 7,Wilcoxon test) more DNA transposons (0.730±0.059% vs.0.472±0.028%,on average 0.55 fold higher) and short interspersed nuclear elements (SINE,0.178±0.008% vs.0.077±0.007%,on average 1.25 fold higher) compared with tinamous.They together contributed an additional 13.98(66.62±7.987 vs.52.64±3.308) Mb,on average,to the ratite genomes compared to those of tinamous (Supplementary Table S5).When inspecting the sequence divergence patterns of DNA transposons compared to their consensus sequences,we did not find any recent expansions in ratites,whose divergence level was expected to be low (Figure 3A;Supplementary Figure S1).Thus,the higher DNA transposon content of ratites is more likely to be caused by the higher rate of DNA transposon removal in tinamous.

Figure 2 Dynamic changes in effective population size

Figure 3 Temporal evolution of transposable elements of paleognaths
In contrast,the predominant repeat type (about 3% of the genome),i.e.,chicken repeat 1 (CR1) LINE elements,showed a similar genomic percentage among paleognaths(Supplementary Table S5).Different CR1 repeat subfamilies exhibited different patterns of genomic composition and temporal activity between the ratites and tinamous.Certain subfamilies,for example,CR1-Y1_Aves and -Y2_Aves,are thought to have been active throughout the evolutionary history of paleognathous species,and probably throughout that of all birds,with previous studies showing activity in both chickens and zebra finches (Suh et al.,2011).This accords with our chronological analyses of nested TEs,with the expectation that younger and active TEs are more likely to be nested within older and inactive TEs (‘transposition in transposition’,TinT (Churakov et al.,2010)) than the opposite scenario.Based on TinT analyses,subfamilies CR1-Y_Pass,-Y4, and -YB2_Pass showed lineage-specific activity throughout the tinamou lineage as well as in some ratites(e.g.,CR1-YB2_Pass in ostriches) (Supplementary Figure S2). This result was corroborated by their sequence divergence patterns compared to their consensus sequences(Figure 3B,Supplementary Figure S3):i.e.,recently active CR1 subfamilies (e.g.,CR1-Y4) were specifically enriched in tinamou species and showed a lower sequence divergence pattern than those (e.g.,CR1-Y2_Aves) active at an earlier time point.Overall,the lower contraction rate of ancient CR1 elements in ratites and more recent expansion of certain CR1 subfamilies in tinamous resulted in the similar level of LINE elements between the two groups of species.
It is worth noting that we also dated two independent horizontal transfers of retroposon AviRTE from filarial nematodes to tinamou species,which is absent in any other paleognaths.One was previously reported in white-throated tinamous (Suh et al.,2016).Based on comparison of the orthologous flanking sequences of AviRTE among species,we inferred another horizontal transfer in the ancestor of little and undulated tinamous within 30 million years (Supplementary Figure S4 and Table S6).
Evolution of gene families
Based on the comparison of orthologous gene families among the 15 paleognathous species,with chickens as the outgroup,we characterized the expanded and contracted gene families across all phylogenetic branches (Figure 4A).We found that genes enriched in the GO terms ‘olfactory receptor activity’,‘G-protein coupled receptor signaling pathway’, or‘spermatogenesis’ frequently exhibited expansion and contraction for both internal and external branches in the Palaeognathae species tree (Dataset 1,2).These immune-or reproduction-related pathways also show rapid turnover in gene families in other species (Demuth et al.,2006; Hahn et al.,2007; Sackton et al.,2007) (Figure 4B,Dataset 1).Interestingly,the ancestor of tinamous showed significant expansions of gene families enriched in GO terms‘oxidoreductase activity’ and ‘developmental maturation’(Figure 4C),which may be related to their small body size,and thus different metabolic rate and developmental speed compared with ratites.Some gene families also showed expansions and were related to sexual development within Tinamiformes:i.e.,‘male genitalia development’,‘testosterone biosynthetic process’,and ‘steroid biosynthetic process’(Dataset 2).Taken together,these results suggest that different species’adaptation processes to different environments and different degrees of sexual selection may have driven changes in gene family size.Interestingly,gene families showing significant contractions in ratites were not enriched in GO terms involved in limb development.This suggests that gene loss is probably not a major force underlying the loss of flight in ratites.
Rapid evolution of mtDNA sequences in birds with highly degenerated W chromosomes
The sex chromosomes of Palaeognathae species were generally less differentiated than those of Neognathae species,with ratite sex chromosomes being even less differentiated than those of tinamous.This may be related to the unique male-only parental care exhibited in paleognathous species,and the low degree of sexual selection targeting males (Wang et al.,2019).To test whether mtDNA genes showed an associated sequence evolutionary pattern with the differentiation degree of sex chromosomes,we calculated the branch-specific cumulative evolutionary rates based on the nonsynonymous to synonymous substitution rate ratio (dN/dS)of 13 mitochondrial genes across the 15 paleognathous birds.We also measured the differentiation degree of sex chromosomes for each species by the length ratio of the Zlinked region that suppressed recombination with the homologous W-linked region (sexually differentiated region,SDR) to the entire Z chromosome.Results demonstrated a significant correlation between the evolutionary rate of mitochondrial genes and sex chromosome differentiation degree (PGLS test,P=0.007 9,Figure 5A).That is,tinamous with a more differentiated sex chromosome pair (a higher SDR/chrZ ratio) tended to fix an excess of slightly deleterious mutations (Supplementary Figure S5) in their mitochondrial genes due to Hill-Robertson interference between the W chromosome and mitochondrial genome.We also found that tinamou mitochondrial genes had a significantly higher(P<2.2e–16,Wilcoxon rank sum test) (Figure 5B) synonymous substitution rate,which evolved largely under neutrality,than that of the ratites.This is consistent with the generally smaller body size and higher basal metabolic rate of tinamous(Figure 5C) compared to ratites.The higher metabolic rate associated with a smaller body size probably has a higher mutagenic effect on mtDNA and nuclear genomes,thus producing a higher mutation rate,as reflected by the synonymous substitution rate.
DISCUSSION
Tinamous are widely distributed throughout the South American continent and occupy different ecological niches,which probably diversified their demographic histories(Figure 2).Here,the trajectories of temporalNechanges were generally different between the ratites and tinamous (Figure 2;Supplementary Table S3).Most species did not show a strong phylogenetic signal regardingNechanges,which may reflect recent population size changes impacted by their separate ecological niches.The closely related Australasian species,i.e.,emus,cassowaries,and kiwis,were the exceptions,demonstrating very similar PSMC patterns despite having different extant geographic distributions (Australia, New Guinea,and New Zealand,respectively).This suggests that their living area used to be largely overlapping.In contrast,the Chilean and elegant-crested tinamous are distantly related but showed very similar population expansions compared to other paleognaths.This may be due to their shared distribution in high-altitude shrubland in southern South America,which may have avoided serious impact from by the LGP.Similarly,white-throated and undulated tinamous shared very similar PSMC patterns and geographic ranges.
Our comparative analyses of the 15 paleognathous nuclear and mitochondrial genomes highlighted that the different sequence evolutionary patterns between ratites and tinamous are likely associated with their different body mass and flight capabilities.
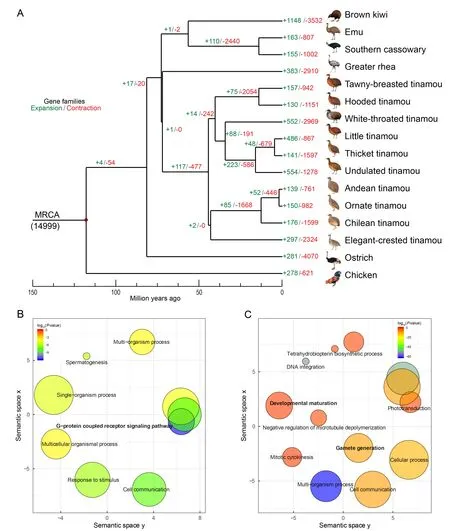
Figure 4 Gene family evolution across Palaeognathae tree

Figure 5 Correlated evolution of mitochondrial genes and W chromosomes among paleognathous species
Differences in TE content in paleognathous species can be impacted by their different effective population sizes (Lynch,2007).Previous studies have demonstrated that recent bottlenecks in endangered mammals can fix excessive TEs in the genome by genetic drift (Abascal et al.,2016; Li & Durbin,2011).Here,population size had a significant association(P<0.05,PGLS test) with overall and certain types of TE content in the studied paleognathous species (Supplementary Table S7),with some exceptions.For example,the dramatic recent expansion of population size in elegant-crested tinamous and contraction of population size in emus have not resulted in dramatically different genome-wide TE content between the two species (5.02% vs.4.98%).However,the recent population size change may impact the frequencies of TE families within the population,as reported inDrosophila(Barrón et al.,2014).All the studied ratites had slightly larger TE content and genome size (Supplementary Tables S1,S5)than that of the studied tinamous.Although species with a smaller estimatedNe(e.g.,emus and cassowaries) may be expected to fix an excess of TEs relative to those with a largerNe(e.g.,ostriches) due to less effective natural selection purging TE insertions (Lynch,2007),all ratites had very similar TE content.Their higher TE content compared with tinamous is consistent with recent comparison between ostriches and white-throated tinamous (Kapusta et al., 2017), which suggested that ostriches,similar to other flightless bird species like penguins,have undergone fewer sequence deletions than flighted birds since their divergence from the common ancestor of birds.Consistent with this pattern,we found that the differences in TE content and genome size between ratites and tinamous were partially attributed to the more severe loss of DNA transposons in tinamous (Figure 3A,Supplementary Figure S1).
Associated with independent loss of flight,gigantism has also evolved multiple times among ratites.We previously showed that ratites have a lower nuclear genome-wide substitution rate than tinamous,which is associated with their body size (Wang et al.,2019).Here,we found a similar pattern for synonymous substitution rates of mitochondrial genes (Figure 5B).This may explain the less differentiated pattern of sex chromosomes in ratites compared with that in tinamous,i.e.,lower rate of genomic rearrangements would suppress homologous recombination between the sex chromosomes.Indeed,a recent comparative analysis of ostrich,falcon,and budgerigar genomes found that ostriches show the lowest genomic rearrangements when all three species are compared to chickens (O’Connor et al.,2018).Among tinamou species with a similar body size,their different estimatedNevalues do not seem to predict the degree of sex chromosome differentiation.For example,in our previous work(Wang et al.,2019),we showed that thicket and little tinamous have very similar patterns and degrees of sex chromosome differentiation,but a nearly 2-fold difference inNeestimated from this work.Interestingly,we found a significant correlation between the extent of sex chromosome differentiation and the mitochondrial gene evolutionary rate.This reflects the effect of Hill-Robertson interference,which would fix slightly deleterious mutations on mtDNA that are genetically linked to the degenerating W chromosome. Tinamous with more degenerated W chromosomes therefore had a higher rate of mitochondria gene evolution than ratites.This could also explain the lower sequence diversity of mitochondria in birds(Berlin et al.,2007) compared to that in mammals,whose mitochondria are not influenced by the paternally inherited Y chromosome.One might expect that strong natural selection on mitochondrial genes due to high metabolic rates could,in turn,retard the degeneration of W chromosomes.However,this does not seem to be the case in hummingbirds:we previously found hummingbirds to have a highly degenerated W chromosome (Zhou et al.,2014),probably because their high metabolic rates evolved after the loss of functional genes on the W chromosome.
CONCLUSIONS
In summary,we delineated the temporal evolution of historical population size,transposable elements,and gene families of paleognathous species in the context of their phylogeny.Their varying degrees of sex chromosome differentiation also allowed us to demonstrate the correlated evolution of maternally-inherited mitochondria and W chromosomes across species,which is expected to only exist in species with female heterogametic sex chromosomes,such as birds and butterflies.
DATA AVAILABILITY
All genomic reads,genome assemblies,and annotations used in this study have been deposited in CNGBdb(https://db.cngb.org/cnsa/) with accession No.CNP0000505.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
Z.J.W.and G.J.C.conducted bioinformatics analyses.Z.J.W.,Q.Z.,and G.J.Z.wrote and revised the manuscript.All authors read and approved the final version of the manuscript.
ACKNOWLEDGEMENTS
We thank China National Genebank at BGI for contributing to the sequencing.We would like to thank Christopher C.Witt,Mariel L.Campbell and Ariel M.Gaffney from the Museum of Southwestern Biology, Gary Graves from Smithsonian Institute,Robb T.Brumfield and Donna L.Dittman from Louisiana State University Museum of Natural Science,Jack Withrow and Andy Kratter from Florida Museum of Natural History,University of New Mexico for providing bird DNA samples for this work.
杂志排行

Zoological Research的其它文章
- The forty-year journey of Zoological Research:advancing with the times
- Chromosomal assembly of the Antarctic toothfish(Dissostichus mawsoni) genome using third-generation DNA sequencing and Hi-C technology
- Geographical range evolution of the genus Polypedates (Anura:Rhacophoridae) from the Oligocene to present
- Molecular and morphological evidence for a new species of the genus Typhlomys (Rodentia:Platacanthomyidae)
- Pitfalls of barcodes in the study of worldwide SARSCoV-2 variation and phylodynamics
- Contribution to the taxonomy of the genus Lycodon H.Boie in Fitzinger,1827 (Reptilia:Squamata:Colubridae) in China,with description of two new species and resurrection and elevation of Dinodon septentrionale chapaense Angel,Bourret,1933