荧光定量PCR用于hTRIMCyPA抑制HIV-1的研究①
2021-03-01吉梦梦孟祥平冯小倩徐欣欣张玉清
吉梦梦 孟祥平 冯小倩 徐欣欣 张玉清 秦 宇
(河南科技大学医学技术与工程学院,洛阳 471000)
HIV-1是反转录病毒科慢病毒属成员,它的基因组是双正链 RNA,主要侵犯人体的CD4+T淋巴细胞和巨噬细胞。当HIV-1进入宿主细胞后,其正链RNA开始反转录合成双链DNA。HIV-1 反转录的过程一般包括以下步骤:①负链强终止DNA(-sssDNA)的合成;②长负链DNA的合成;③正链强终止DNA(+sssDNA)的合成;④完整的双链DNA合成[1]。最后合成的完整DNA两端含有LTR序列,又叫HIV-1晚期反转录产物(HIV-1 late RT),因此在检测反转录过程完整的 DNA双链时,应当检测 LTR/Gag序列[2]。反转录过程是HIV-1病毒复制的关键环节,已成为研发抗HIV-1药物的重要靶点[3]。恒河猴TRIM5α属于三重基序蛋白(tripartite motif protein,TRIM)大家族的成员,通过多种机制抑制HIV-1,而人的TRIM5α作用较弱[4-8]。将人TRIM5α中结合衣壳蛋白的B30.2(SPRY)结构域用同样结合衣壳蛋白的亲环素A(CyPA)代替,构建人源的hTRIMCyPA以避免异种蛋白的免疫排斥反应。因此推测hTRIMCyPA也能特异识别并结合HIV-1衣壳蛋白,并影响衣壳蛋白的脱壳,抑制病毒的RNA反转录成DNA[4-5]。虽然临床有效的抗反转录病毒治疗(HAART)提高了艾滋病的临床疗效,但因HIV-1的高度变异性和潜伏感染,存在药物疗效不佳和毒副作用大的问题[9-10]。因此,本研究采用假病毒系统建立一种可分析感染细胞内病毒生命周期早期特异性DNA 产物的相对荧光定量 PCR技术,并以其评价hTRIMCyPA对HIV-1的抑制作用。这对于未来抗HIV-1相关病毒新药的研发具有重要的意义。
1 材料与方法
1.1材料 蛋白酶K、DNase Ⅰ购自Sigma公司;SYBR Green Master Mix(Low Rox)购自上海翊圣生物科技有限公司;EZTrans细胞转染试剂购自上海李记生物;荧光素酶检测试剂盒购自碧云天生物技术公司;兔抗GFP和内参GAPDH多克隆抗体购自上海生工生物公司;hTRIMCyPA基因表达质粒hTRIMCyPA/pcDNA3.1EGFP由本实验室构建,pNL4-3LucR-E-由徐建青教授惠赠,HEK293细胞、HEK293t细胞、pMD2.G、pcDNA3.1和pcDNA3.1EGFP载体由本实验室保存。
1.2方法
1.2.1PCR引物的设计与合成 引物由premier5.0软件设计,由上海生工生物公司合成。HIV-1晚期反转录产物基因引物的设计:参考 pNL4-3LucR-E-的序列,选择HIV-1的LTR区和HIV-1Gag之间的序列设计晚期反转录产物引物,设计合成引物如下:LTRGag F:5′-TGTGTGCCCGTCTGTTGTGT-3′,R:5′-GAGTCCTGCGTCGAGAGATC-3′。作为内参基因的GAPDH基因引物如下:GAPDH F:5′-CAAGGCTGTGGGCAAGGTC-3′,R:5′-TGGAGGAGTGGGTGTCGCT-3′。
1.2.2HIV-1假病毒包装 在包装的前一天进行铺板,待HEK293T细胞生长至约80%汇合度时,将质粒pMD2.G和pNL4-3LucR-E-以1∶3的比例联合转染到细胞中,转染按照 EZTrans细胞转染试剂说明书操作进行。转染6 h后换液,48 h和72 h各收集全部上清,800 g离心5 min以去除细胞碎片,用0.45 μm的滤器过滤,分装后保存备用。
1.2.3hTRIMCyPA的表达及Western blot鉴定 将hTRIMCyPA/pcDNA3.1EGFP及pcDNA3.1转染HEK293细胞,第二天在荧光显微镜下观察hTRIMCyPA的表达,48 h后,吸去培养液,用1 ml预冷的PBS洗1次,吸去 PBS,再用碧云天的IP及Western细胞裂解液(含1 mmol/L PMSF)裂解并收获细胞,蛋白样品经12%SDS-PAGE后,转移至PVDF 膜,封闭后,5%脱脂牛奶封闭1 h后,分别加入Anti-EGFP 及内参GAPDH多抗4℃孵育过夜,洗膜后加入偶联HRP的羊抗兔二抗,室温孵育2 h,洗膜后进行ECL显色。
1.2.4假病毒感染实验 将1.2.3转染hTRIMCyPA/pcDNA3.1EGFP及pcDNA3.1的细胞作为感染靶细胞,每组3个复孔。转染前弃去培养基,每孔加入500 μl含2 ng p24 的假病毒,假病毒事先在37℃下10 mmol/L MgCl2中的用DNase Ⅰ(20 U/ml)预处理病毒1 h以除去残存的质粒DNA。为提高感染效率,加入1 μg/ml的polybrene。6 h后,用PBS洗3次以去除残留未感染的假病毒。再继续培养至48 h,使用荧光素酶检测试剂盒检测荧光素酶活性以确认是否感染成功。
1.2.5细胞中总DNA的提取1.2.4操作至用 PBS 洗3 次去除残留未感染的假病毒后,在24孔板中直接加入600 μl细胞裂解液(1.5 mmol/L pH8.0 Tris-HCl,50 mmol/L KCl,1.5 mmol/L MgCl2,0.45%Np-40,0.45%Tween20)充分裂解,混匀后加入蛋白酶K(终浓度为100 μg/ml),55℃过夜或57℃ 3 h,95℃15 min水浴灭活蛋白酶K,4℃,12 000 g 离心5 min取上清作为待测样品。
1.2.6SYBR Green Ⅰ荧光定量PCR方法建立 按照SYBR Green Ⅰ荧光PCR试剂盒说明书操作,20 μl反应体系:2×SYBR Green Ⅰ Mixture 10.0 μl,10 μmol/L Forward Primer 0.4 μl,10 μmol/L Reverse Primer 0.4 μl,Template DNA 1 μl,加ddH2O至20 μl。循环条件:95℃ 5 min;95℃ 10 s;58℃ 40 s,40个循环;58℃ 1 min退火阶段采集荧光信号。绘制标准曲线:取hTRIMCyPA组所提取的基因组DNA进行2倍梯度稀释,做7个梯度,分别用内参引物和目的基因引物进行扩增。仪器自动绘制出对应于内参基因和目的基因的标准曲线。所有试验样品均设3次重复。采用 2-ΔΔCt计算HIV-1反转录产物的相对表达量,用内参基因校准目的基因表达。

2 结果
2.1hTRIMCyPA的表达及Western blot鉴定 将hTRIMCyPA/pcDNA3.1EGFP和pcDNA3.1空载体质粒用EZTrans细胞转染试剂转染HEK293细胞。第二天用荧光显微镜观察绿色荧光蛋白表达情况,发现转染hTRIMCyPA孔出现明显的绿色荧光(见图1B),而对照孔没有绿色荧光表达(见图1A)。hTRIMCyPA与载体中的EGFP融合,因此可以判断hTRIMCyPA表达成功。为进一步确定该蛋白的表达,将hTRIMCyPA/pcDNA3.1EGFP或pcDNA3.1空载体质粒转染试剂转染HEK293细胞,48 h后裂解细胞进行Western blot检测,由于hTRIMCyPA与EGFP融合,因而hTRIMCyPA对应的孔出现条带而空载体组及空白细胞对照组均没有条带(见图2),进一步说明hTRIMCyPA成功表达。
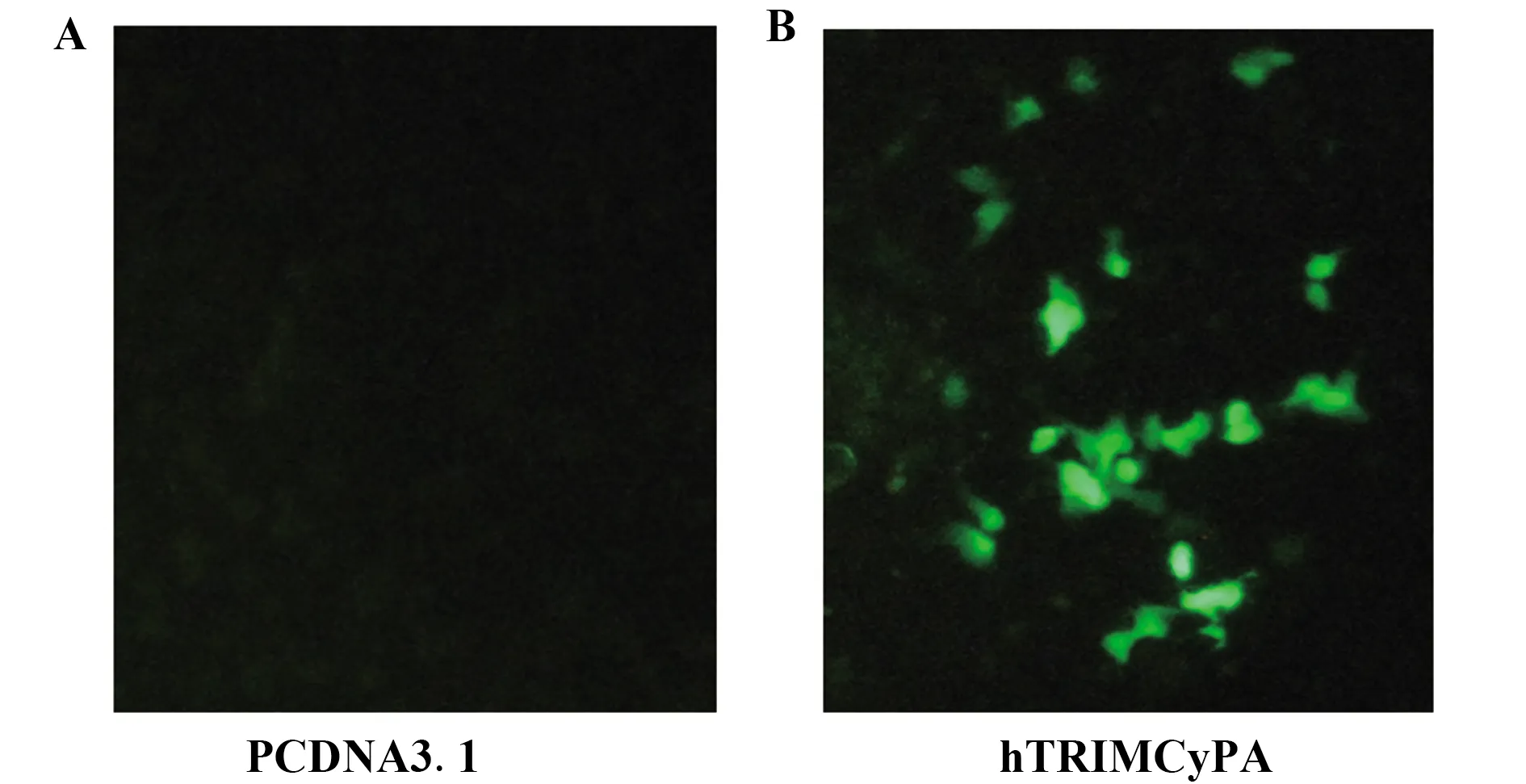
图1 hTRIMCyPA表达的荧光显微镜检测
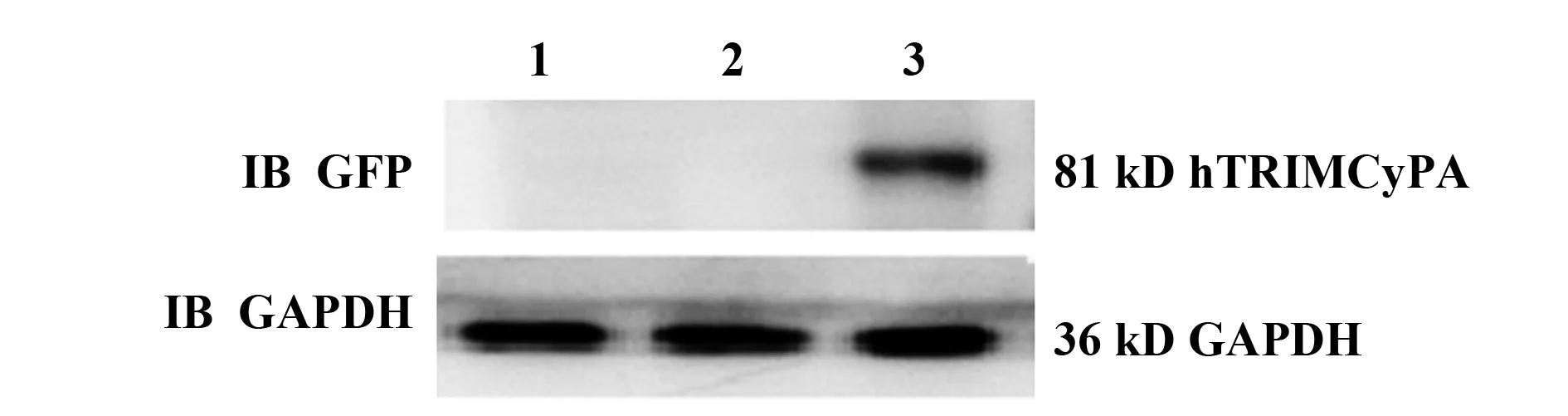
图2 hTRIMCyPA的Western blot鉴定图
2.2假病毒感染 荧光素酶检测试剂盒检测荧光素酶活性,结果发现转染后2组的荧光素酶活性明显高于空白细胞对照组。hTRIMCyPA组的荧光素酶活性低于空载体组转染组(见图3),说明HIV-1 假病毒颗粒感染成功,并且hTRIMCyPA具有抑制HIV-1的作用。
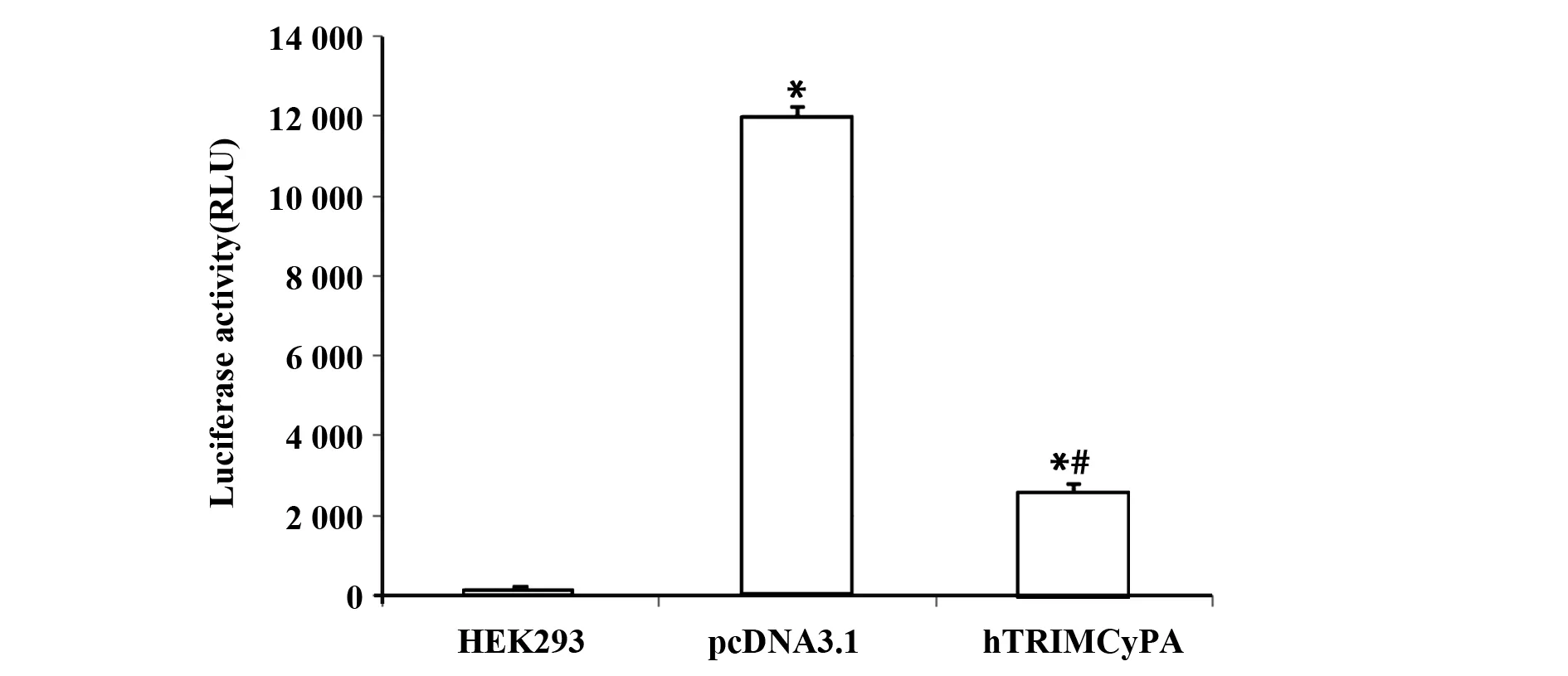
图3 假病毒感染HEK293细胞后的荧光素酶活性分析
2.3荧光定量PCR检测体系的建立 在QuantStu-dio 3 型荧光实时PCR检测仪上结束扩增后,由系统软件分析,以初始模板的对数为x轴,以Ct值为y轴作回归曲线,自动生成标准曲线。其中HIV-1 late RT标准曲线(见图4A)的相关系数R2为0.992 9。斜率为-3.381,标准曲线回归方程为:y=-3.381x+27.208。内参GAPDH标准曲线(见图4B)相关系数R2为0.993 7。斜率为-3.289,标准曲线回归方程为:y=-3.289x+25.307。二者的斜率相差低于0.1,表明二者扩增效率一致,可用于接下来的相对定量分析。相关系数R2>0.99,表明两标准曲线线性较好,能够准确定量。

图4 HIV-1 late RT 和 GAPDH 基因标准曲线
2.4各样品相对定量分析结果 利用本试验建立的SYBR GreenⅠ荧光定量 PCR方法对假病毒感染的hTRIMCyPA表达的样本组的抑制效果见图5。可见hTRIMCyPA组HIV-1相对late RT量明显少于空载体组,有统计学意义。
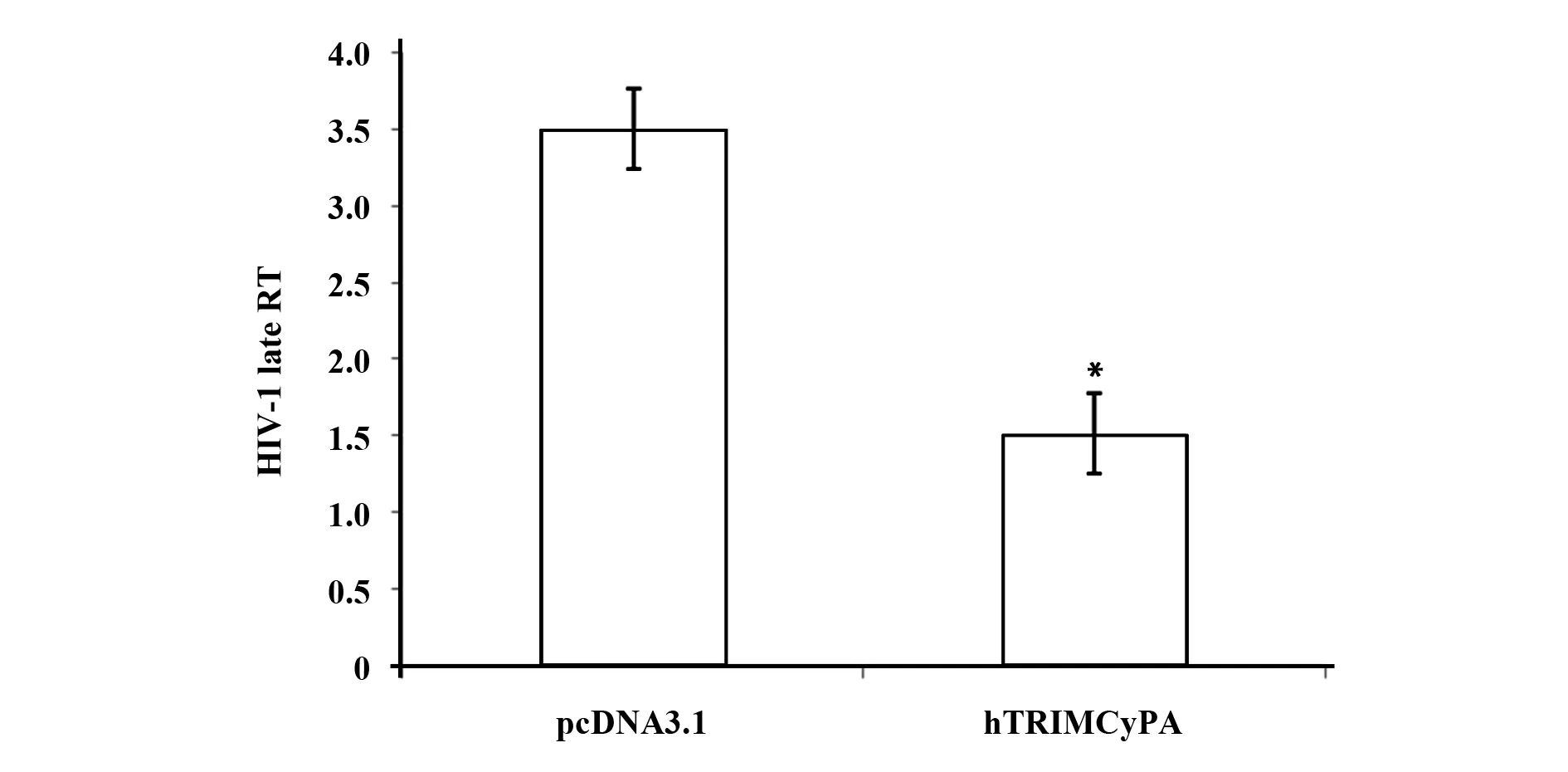
图5 HIV-1假病毒感染后各组HIV-1 late RT产物的相对量
3 讨论
荧光定量PCR克服了常规 PCR 只能定性不能定量的技术难题,其包括绝对和相对两种定量方法。绝对定量法需要克隆相关的基因以构建相应表达载体作为标准品用于构建标准曲线,并且每次试验均要制作标准曲线,增加了实验的复杂度。本研究以 SYBR GreenⅠ作为荧光标记物,不需要设计昂贵的探针,因而成本相对较低。为了验证目的基因和内参基因扩增的Ct值与拷贝数的相关性,本研究建立了各自标准曲线加以验证。在实时定量中,只要将待测样品中的任何一个作为 “相对标准品”,系列稀释后用于构建标准曲线,即可获得斜率并验证方法的可行性[11-12]。这无疑降低了实验的工作量和复杂性。
本研究所设计的HIV-1 late RT引物,上游处于LTR序列,下游处于Gag前端,扩增出LTR/Gag序列代表连续完整的HIV-1序列。若把引物设计在Gag或Luc等部位,扩增出来的片段不能够代表完整片段,很可能包含一些中间片段,部分是中间合成的长负链 DNA产物。本次研究也曾用Gag或Luc等部位设计的引物进行扩增,发现其PCR产物的量明显大于LTR/Gag引物扩增的量,原因是这个引物扩增的产物含有很多不完整片段,因而不能真正反映完整HIV-1反转录产物量,也不能代表HIV前病毒量[2,13]。据报道,ZACK等[14]利用 PCR检测受激和静息状态人类T淋巴细胞中 HIV-1的反转录过程时,发现R/U5序列、U3序列和U3/R/U5-PBS之间序列所扩增出的代表初始反转录片段产物数量几乎相同,但受激T细胞中LTR和Gag之间扩增出的代表完整 DNA片段数量明显多于静息细胞,所以选择扩增的LTR/Gag连续序列,能够比较真实地反映HIV-1的反转录情况及病毒活性。
对于HIV-1DNA的提取一般参考基因组的提取方法,一般采用带有DNA吸附柱的试剂盒提取或酚/氯仿抽提DNA法提取[15-16]。这2种方法一般要经过多次转管和离心沉淀,但是HIV-1转录后不会马上整合到庞大而复杂的染色体上,有一部分最先还是些非整合的线形或环状双链DNA分子,通常称为赫特(Hirt)DNA,因此有特定的提取方法[17-18]。在经过多次转管和离心沉淀后,这些散在的DNA可能并不一定和染色体基因组同比例的丢失,因而容易造成DNA得率较低以及实验的重复性差。而直接用细胞裂解液处理后直接PCR在一定程度上能避免这个问题,同时也降低了实验的工作量。目前直接PCR已经在基因组的PCR扩增中得到了广泛的应用,与用试剂盒提取以及其他方法相比,无论是扩增质量还是效率等都具有明显的优势[19-20]。
本研究所用的pNL4-3LucR-E-HIV-1假病毒载体,因其基因序列中的膜蛋白基因被失活,需要与膜蛋白基因质粒共转染才能包装出子代假病毒,由此包装的假病毒由于仅具有单次感染能力而十分安全;另外,由同一外源质粒表达所提供的膜蛋白可产生均一的假病毒颗粒,因而实验的可重复性得到保证,这样就避免了HIV-1活病毒反复传代变异而导致的重复性差的问题[21]。有研究报道,用假病毒来进行药物筛选与同时用活病毒筛选结果一致,从而大大提高了药物筛选的通量和安全性[22-23]。利用本研究建立的方法已经成功筛选了多个抗病毒药,并对其机制进行了研究。因而本研究建立的测定HIV-1特异性反转录产物的荧光定量PCR的方法具有简单、高效和成本低廉等优点,可用于抗病毒药物机制研究和药物筛选。
致谢:感谢河南科技大学大学医学技术与工程学院段万理、张克兰和秦怡帆同学,河南科技大学大学医学院马甜和余冯同学在实验过程中给予的帮助。
