绿色荧光蛋白分裂肽基因重组乙型肝炎病毒复制子模型的建立
2021-03-01田青右朱园飞常豪于琳邓强
田青右,朱园飞,常豪,于琳,邓强
复旦大学上海医学院基础医学院病原生物学系, 教育部/卫健委/医科院医学分子病毒学重点实验室,上海 200032
乙型肝炎病毒(hepatitis B virus, HBV)是一类有包膜的小型DNA病毒,属于嗜肝DNA病毒科[1]。成熟的HBV含有约3.2 kb的松弛环状DNA(relaxed circular DNA, rcDNA),负链DNA 5′端共价连接病毒多聚酶(polymerase,Pol)。在病毒感染的肝细胞核内,rcDNA修复为共价闭合环状DNA(covalently closed circular DNA, cccDNA),是HBV转录复制的原始模板[2-4]。以cccDNA为模板转录的前基因组RNA(pregenomic RNA, pgRNA)转运至细胞浆被核衣壳包裹,通过Pol反转录生成负链DNA,并合成不完全的正链DNA;部分双链rcDNA通过成熟的核衣壳颗粒再次转运至细胞核,完成HBV的细胞内复制周期[5]。另一方面, HBV核衣壳颗粒通过内体分选转运复合体机制,经由内体多泡体结构完成包膜化和出芽。HBV已知的天然宿主仅限于人和黑猩猩,钠牛磺胆酸共转运多肽(sodium taurcholate cotransport peptide, NTCP)被证明是HBV高亲和力受体[6-8],这极大地促进了研究者对HBV从头感染的研究。然而,HBV/NTCP感染体系依赖较高的感染复数,病毒感染和扩增效率较低,病毒粒子表面抗原与受体结合后的细胞进入机制仍需要深入研究。
重组病毒复制子通常在病毒基因组或亚基因组中插入外源报告基因,使其能够支持重组病毒进行功能性复制,具有可检测性和量化特征,如在丙型肝炎病毒、新型冠状病毒[9-13]RNA复制子中引入荧光素酶或荧光蛋白表达基因等,是病毒学体外研究的重要工具。值得注意的是,HBV 3.2 kb基因组高度压缩,4个主要阅读框相互重叠,每一个碱基都具有编码功能和潜在的顺式调控功能,因此HBV基因组难以容纳外源基因,构建重组复制子具有较大的挑战性。Hong等[14]应用包膜蛋白缺陷的HBV高复制突变株,在病毒Pol spacer编码区插入红色荧光蛋白(red fluorescent protein, RFP)基因,能够支持HBV重组病毒(recombinant HBV, rHBV)的复制;Bai等[15]基于上述突变株系统重新设计rHBV复制子,将外源报告基因插入HBV核心蛋白(HBV core, HBc)编码区,通过反式互补HBc拯救重组病毒复制。尽管如此,受限于大片段插入导致的低复制效率,上述系统并未能获得广泛应用。
内含肽(intein)可分割为氮端(IntN)和碳端(IntC)结构域[16-18],应用IntN、IntC分别融合绿色荧光蛋白(green fluorescent protein, GFP)氮端(GFPN)和碳端(GFPC)分裂蛋白,通过IntN和IntC高亲和力相互作用、催化自身移除,介导GFP分裂蛋白基于肽键的共价连接。
鉴于此,本研究基于HBV ayw3通用基因组序列,删除HBc第23~136氨基酸(amino acid, aa)编码区,构建复制缺陷的ΔHBc113载体。由于插入序列容量限制,选取intein融合的GFP作为报告基因产物。依据11个β-片层组成的二级结构,将增强绿色荧光蛋白(enhanced GFP, EGFP)分割成EGFPN1-8(aa 1~173)和EGFPC9-11(aa 174~232);或将超折叠绿色荧光蛋白(super folder GFP, sfGFP)拆分为sfGFPN1-10(aa 1~214)和sfGFPC11(aa 215~230)。应用IntC融合EGFPC或sfGFPC,将其序列插入ΔHBc113载体,在HBc反式互补条件下实现重组病毒rHBV的复制,IntC-GFPC表达产物则能够与共表达的IntN-GFPN高效拼接,产生功能性荧光蛋白。期望这一可视化的模型系统能为HBV高通量药物筛选提供实验工具,也有助于对HBV受体和受体后病毒学特征的进行深入研究。
1 材料与方法
1.1 材料
1.1.1 细胞系293T细胞系、HepG2细胞系由本课题组保存。
1.1.2 质粒pCDNA3.1、巨细胞病毒(cytomegalovirus, CMV)-HBV1.1(由CMV启动子启动的1.1拷贝的HBV基因组)、pCDH-CMV-MCS-EF1-puro(由CMV启动子启动、带有多克隆位点和筛选标记Puro的慢病毒载体)、pCDH-EGFP、pCDH-sfGFP均由本课题组保存。pCDNA3.1-IntC-EGFPC9-11(可表达intein碳端和EGFP碳端的融合蛋白)、pCDNA3.1-EGFPN1-8-IntN(可表达EGFP氮端和intein氮端的融合蛋白)、pCDNA3.1-IntC-sfGFPC11(可表达intein碳端和sfGFP碳端的融合蛋白)、pCDNA3.1-sfGFPN1-10-IntN(可表达sfGFP氮端和intein氮端的融合蛋白)均由上海桑尼生物科技有限公司合成。
1.1.3 仪器和试剂主要仪器有聚合酶链反应(polymerase chain reaction,PCR)仪(Eppendorf公司)、二氧化碳细胞培养箱(Thermo 公司)、荧光倒置显微镜(Nikon公司)、pH计(上海天达仪器有限公司)、化学发光成像系统ChemiDocXRS+(Bio-Rad公司)。主要试剂有PrimeSTAR Max DNA Polymerase(TaKaRa公司)、Seamless Cloning试剂盒(碧云天生物技术有限公司)、无内毒素质粒小提中量试剂盒[天根生化科技(北京)有限公司]、凝胶DNA小量回收试剂盒(美基生物有限公司)、EZ Trans细胞转染试剂(上海李记生物科技有限公司)、DMEM培养基(Gibco公司)、胎牛血清(Gibco公司)、Trypsin-EDTA(Gibco公司)。所用的引物由北京擎科新业生物技术有限公司合成,引物序列见表1。

表1 PCR引物
1.2 方法
1.2.1 质粒构建以pCMV-HBV1.1(HBV ayw3,GenBank N°V01460)为模板,ΔHBc113-F和ΔHBc113-R为引物进行PCR,利用Seamless Cloning试剂盒,将PCR产物无缝连接在pCMV-HBV1.1载体NdeⅠ和BspEⅠ酶切位点之间,获得HBV1.1-ΔHBc113质粒。
通过基因合成获得念珠藻类(NostocpunctiformePCC73102, Npu)intein编码序列。应用常规PCR扩增获得IntC-EGFPC9-11、EGFPN1-8-IntN、IntC-sfGFPC11以及sfGFPN1-10-IntN融合蛋白编码DNA,分别克隆至真核表达质粒pCDNA3.1(+)。以相应质粒为模板,应用IntC-EGFPC9-11-F和IntC-EGFPC9-11-R,或IntC-sfGFPC11-F和IntC-sfGFPC11-R为引物分别进行PCR扩增,将PCR产物连接在HBV1.1-ΔHBc113载体NdeⅠ和BspEⅠ酶切位点之间,构建质粒分别命名为ΔHBc113/EGFPC9-11和ΔHBc113/sfGFPC11。以EGFPN1-8-IntN-F和EGFPN1-8-IntN-R,或sfGFPN1-10-IntN-F和sfGFPN1-10-IntN-R为引物,分别扩增EGFPN1-8-IntN和sfGFPN1-10-IntN融合蛋白编码序列,将PCR产物克隆至pCDH-puro载体EcoRⅠ和BamHⅠ酶切位点间,构建慢病毒质粒pCDH-EGFPN1-8-IntN和pCDH-sfGFPN1-10-IntN。
1.2.2 细胞培养和细胞转染将HepG2和293T细胞置于5% CO2培养箱37 ℃培养,培养基为DMEM(含10%胎牛血清、100 U/mL青霉素和 100 μg/mL 链霉素)。将处于生长对数期的HepG2或293T细胞接种到六孔板中,24 h后细胞密度达到80%~90%准备转染。将质粒按每孔3 μg总量加至125 μL DMEM无血清培养基中,另外将9 μL EZ Trans细胞转染试剂加至125 μL DMEM无血清培养基中,充分混匀DNA溶液和转染试剂溶液,静置20 min。弃掉细胞培养液,换入2 mL新鲜无血清DMEM培养基。将质粒脂质体混合液加入细胞中,轻轻混匀后放入细胞培养箱培养。6~8 h后,弃掉培养液,加入新鲜含血清DMEM培养基,放入培养箱中培养,以供后续实验。
1.2.3 荧光信号检测实验HepG2和293T细胞经EZ Trans转染试剂转染质粒24 h后,弃去培养基,每孔加入2 mL新鲜的含血清DMEM培养液,放入培养箱中继续培养。24 h后,在荧光倒置显微镜下观察细胞绿色荧光信号,拍照记录结果。
1.2.4 提取HBc颗粒内DNAHepG2细胞转染质粒3~4 d后,弃上清,用预冷的磷酸盐缓冲液(phosphate buffered saline, PBS)洗2次。六孔板每孔加入400 μL裂解液,在37 ℃培养箱放置 30 min。将裂解液 14 000g(下同)离心5 min,弃沉淀物,收集上清液,加入4 μL 1 mol/L MgCl2和8 μL 10 mg/mL DNase Ⅰ,37 ℃水浴消化30 min。然后用100 μL 35% PEG 8 000 沉淀病毒颗粒,4 ℃过夜放置,离心10 min,沉淀用100 μL DNase Ⅰ溶液重悬,37 ℃水浴消化30 min。用蛋白酶K消化去除病毒核衣壳,37 ℃水浴消化过夜。用等体积的苯酚抽提1次,振荡后离心10 min,取上清水相,加入 2 μL 20 mg/mL糖原、十分之一体积3 mol/L醋酸钠(pH 5.2)溶液和等体积的异丙醇,混匀后置于 -20 ℃沉淀过夜,离心15 min弃上清,沉淀用75%乙醇洗2次并尽弃乙醇,静置待乙醇挥发,用20 μL去离子水溶解,-80 ℃保存。
1.2.5 提取细胞上清HBc颗粒内DNAHepG2细胞经EZ Trans转染试剂转染质粒24 h后,收集培养上清液, 2 000g离心5 min,去除细胞碎片,用孔径0.45 μm的滤膜过滤。加入PEG 8 000 溶液至终浓度7%,4 ℃放置过夜。取浓缩后的400 μL上清液,重复 1.2.4 步骤,提取HBc颗粒内DNA。
1.2.6 DNA印迹法(Southern blotting)检测rHBV载体的细胞内复制能力配制160 mL 1%琼脂糖凝胶,加入抽提的HBV胞内和上清DNA样品, 80 V电泳 3 h。电泳结束后,将凝胶用变性液洗2次,每次 15 min,用去离子水润洗后,再用中性液中和2次,每次15 min。将凝胶再次用去离子水清洗后,放入20×SSC浸泡。将尼龙膜、滤纸完全浸湿,按照吸水纸、滤纸、尼龙膜、凝胶、滤纸由下至上的顺序进行搭桥过夜。转膜结束后,取出尼龙膜,用2×SSC浸泡5 min,再置于2张滤纸之间,于80 ℃烘烤2 h。尼龙膜放入杂交管,用10 mL预杂交液浸润,在杂交炉42 ℃预杂交30 min。倒出预杂交液,将预热好的探针放入杂交管中,42 ℃旋转过夜孵育。回收探针,尼龙膜用10 mL高盐溶液常温洗2次,每次15 min,接着用预热的68 ℃低盐溶液洗2次,每次15 min。洗液润洗尼龙膜2 min后用吸水纸吸干,加入封闭液室温孵育30 min,再用anti-DIG antibody溶液室温孵育30 min。洗液室温润洗尼龙膜3次,每次10 min。先用显色液室温平衡膜2~5 min,加入CSPD显色液避光常温孵育5 min后,用化学发光检测仪器检测,记录结果。
2 结果
2.1 基于HBV1.1-ΔHBc113载体的重组病毒复制子的构建
基于HBV ayw基因型通用序列,设计构建HBc缺失的HBV复制子载体HBV1.1-ΔHBc113(图1A)。在HBc表达质粒共转染条件下,HBV1.1-ΔHBc113载体显示出良好的HBV DNA细胞内复制信号(图1B)。由于HBV1.1-ΔHBc113载体HBc部分序列被删除,所以与野生型相比,HBV1.1-ΔHBc113复制子形成的rcDNA和双链线性DNA(double-stranded linear DNA, dslDNA)条带更靠前。基于上述载体进一步构建携带外源报告基因的重组复制子(图1C),由独立的ATG位点翻译起始外源基因的表达,3′端则设计T2A断裂肽引导HBV Pol蛋白的表达。然而,引入全长GFP、RFP以及荧光素酶等报告基因,都未能有效支持重组病毒复制。
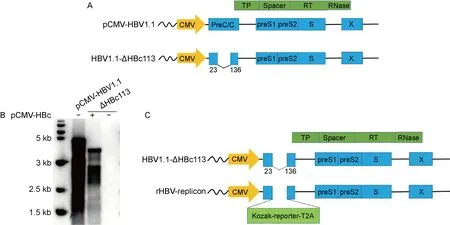
A: Schematic representation of HBV1.1 replicon and ΔHBc113 mutant. B: Southern blotting of HBV intracellular replication. ΔHBc113 vector was transfected with or without pCMV-HBc in HepG2 cells. HBV1.1 replicon was served as a positive control. C: Schematic diagram of rHBV construct bearing a reporter gene. The foreign gene is initiated from an optimized Kozak signal. An in-frame T2A coding sequence is designed to facilitate HBV Pol gene expression.
2.2 EGFPC9-11/EGFPN1-8分裂肽在intein作用下形成完整GFP
Intein分割为IntC和IntN, IntC和IntN侧翼分别融合GFPN和GFPC分裂蛋白,通过IntN和IntC高亲和力相互作用、催化自身移除,介导GFP分裂蛋白基于肽键的共价连接(图2A)。首先将EGFP分成aa 1~173(EGFPN1-8)和aa 174~232(EGFPC9-11)两部分(图2A),基于pCDNA3.1和pCDH-puro质粒,设计构建EGFPC9-11-IntC和EGFPN1-8-IntN真核表达载体。结果显示,在体外培养的293T细胞中,单独转染EGFPC9-11-IntC或EGFPN1-8-IntN质粒未能观察到绿色荧光;当共转染2个互补质粒时,则能够观察到显著的绿色荧光信号,但弱于完整EGFP质粒转染细胞的荧光强度(图2B)。在缺乏intein基序融合时,EGFPC9-11/EGFPN1-8表达质粒共转染,同样能够诱导GFP产生,但荧光强度显著减弱(图2C)。
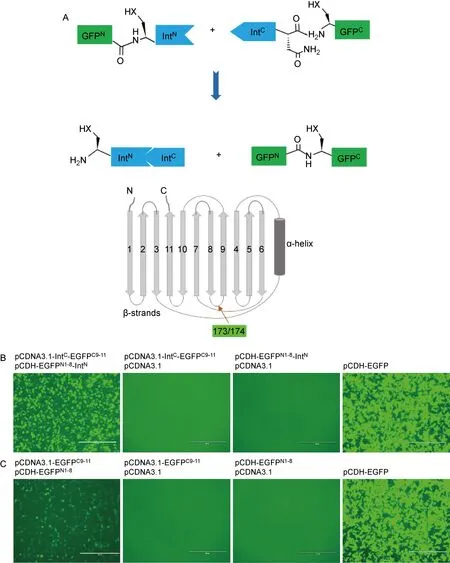
A: Schematic diagram of split-GFP through intein trans-splicing pathway and consecutive β-strand numbering was shown in the EGFP structure diagram. Arrow indicates the split site (between aa 173 and 174). B: 293T cells transfected with pcDNA3.1/pCDNA3.1-IntC-EGFPC9-11, pcDNA3.1/pCDH-EGFPN1-8-IntN, or pCDNA3.1-IntC-EGFPC9-11/pCDH-EGFPN1-8-IntN, respectively. pCDH-EGFP was served as control. Scale bars, 400 μm (the same below). C: Green fluorescence signals reduced greatly in split-EGFPN1-8/C9-11 system in the absence of intein-mediated splicing.
2.3 sfGFPC11/sfGFPN1-10分裂肽在intein作用下形成完整GFP
为获得较短的GFP碳端分裂肽,本研究尝试应用sfGFP,即所谓精良折叠版本的GFP(其对融合蛋白结构具有更好的耐受性)。将sfGFP拆分为aa 1~214(sfGFPN1-10,214 aa)和aa 215~230(sfGFPC11,16 aa)(图3A),共转染实验结果显示sfGFPC11-IntC与sfGFPN1-10-IntN具有良好的匹配,能够诱导高效拼接并产生高亮度荧光蛋白(图3B)。同样,sfGFPC11-IntC与sfGFPN1-10-IntN质粒单独转染均不能产生荧光信号,sfGFPC11/sfGFPN1-10的拼接效率显著弱于sfGFPC11-IntC/sfGFPN1-10-IntN(数据未展示)。
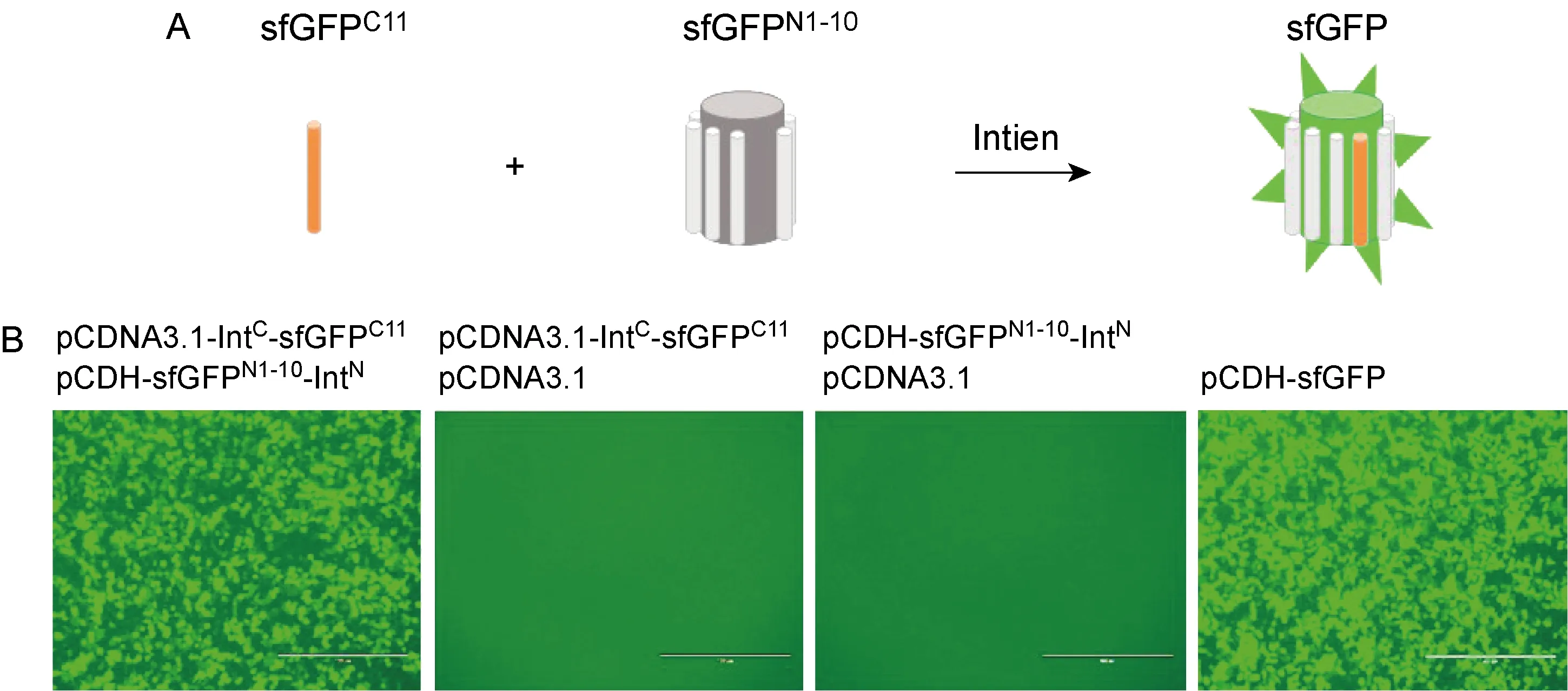
A: Schematic representation of split-sfGFP system. B: 293T cells transfected with pcDNA3.1/pCDNA3.1-IntC-sfGFPC11, pcDNA3.1/pCDH-sfGFPN1-10-IntN, or pCDNA3.1-IntC-sfGFPC11/pCDH-sfGFPN1-10-IntN, respectively. pCDH-sfGFP was served as control. Scale bars, 400 μm.
2.4 编码GFP分裂肽的重组ΔHBc113载体能够在HBc互补条件下有效复制
将IntC-EGFPC9-11或IntC-sfGFPC11编码序列分别插入ΔHBc113载体,基于图1 C构建ΔHBc113/EGFPC9-11和ΔHBc113/sfGFPC11两种rHBV复制子。如图4A所示,在肝肿瘤细胞来源的HepG2细胞中,应用两种rHBV复制子载体与相应的EGFPN1-8-IntN或sfGFPN1-10-IntN表达质粒共转染,观察到了荧光,证明成功诱导了功能性GFP的拼接。
在HepG2转染细胞中,应用Southern blotting验证两种rHBV载体的细胞内复制能力。如图4B所示,单独转染ΔHBc113/EGFPC9-11或ΔHBc113/sfGFPC11质粒均不能诱导重组病毒细胞内复制。在共转染HBc表达质粒的条件下,ΔHBc113/EGFPC9-11仅显示非常弱的病毒DNA复制中间体信号,外源基因较短的ΔHBc113/sfGFPC11病毒复制信号则显著增加,但仍弱于ΔHBc113空载体。进一步用酶切方式验证重组病毒复制中间体,如图4B所示,由于rcDNA部分双链序列中含有EcoRⅠ限制酶切位点,酶切处理后形成3.2 kb (电泳位置)的线性DNA,提示ΔHBc113/sfGFPC11载体具有可靠的细胞内复制能力。
将ΔHBc113/sfGFPC11和pCMV-HBc共转染HepG2细胞,通过PEG 8000沉淀、浓缩培养细胞上清。将pCMV-HBV1.1转染细胞上清作为阳性对照,单独转染ΔHBc113/sfGFPC11载体作为阴性对照,Southern blotting证实ΔHBc113/sfGFPC11重组复制子能够在HBc回补条件下产生子代病毒粒子(图4C)。
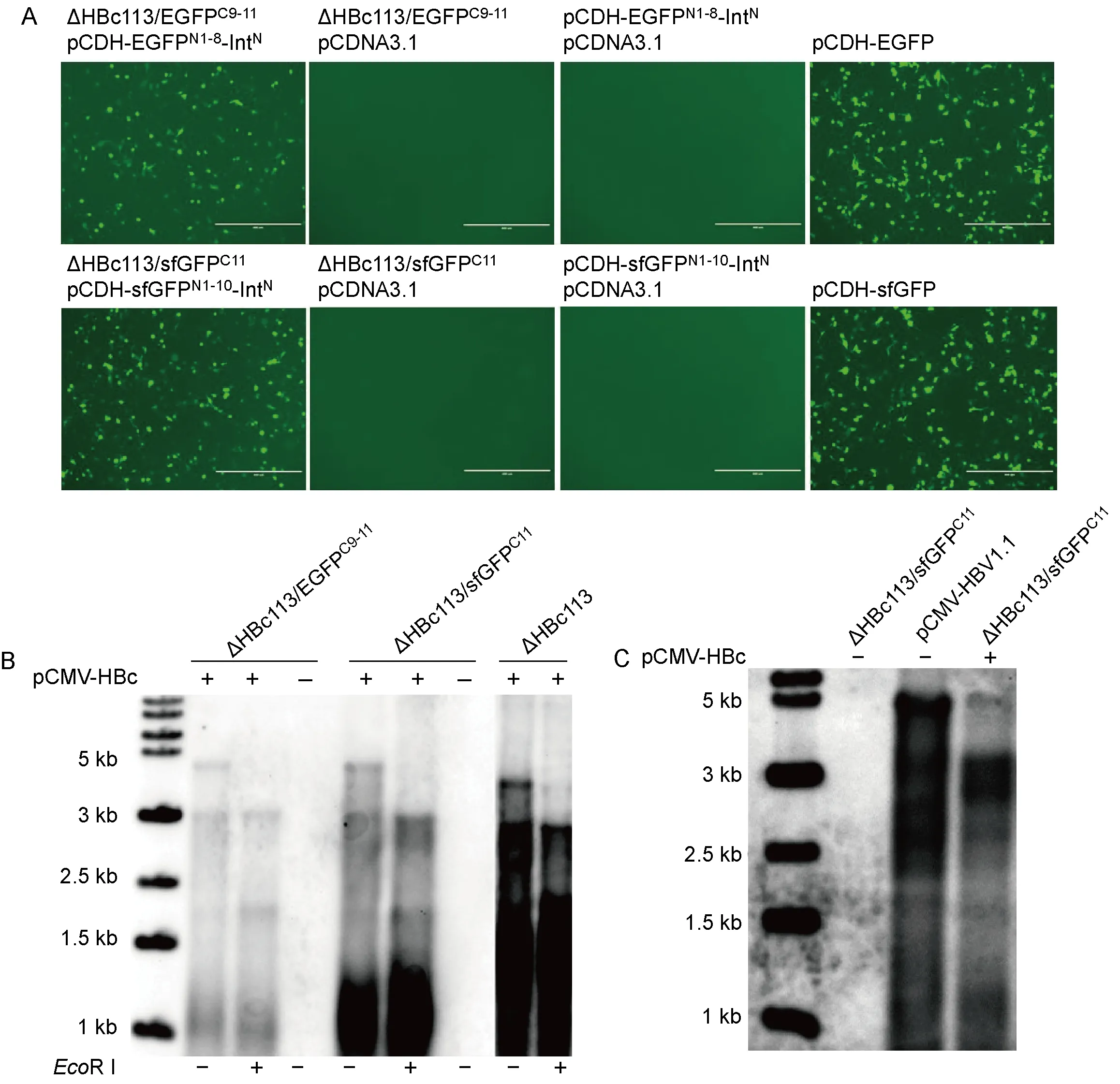
A: Fluorescence analysis of HepG2 cells co-transfected with ΔHBc113/EGFPC9-11 or ΔHBc113/sfGFPC11, and their counterpart plasmids as indicated, respectively. Scale bars, 400 μm. B: Southern blotting of HepG2 cells transfected with ΔHBc113/EGFPC9-11or ΔHBc113/sfGFPC11, with or without pCMV-HBc co-transfection. rHBV DNA intermediates were validated by EcoR Ⅰ digestion. C: HepG2 cells were transfected with indicated plasmids. Virions concentrated from cell culture supernatants were subjected to Southern blotting.
3 讨论
本研究实际包含两个层次的蛋白分裂系统,即GFP分裂系统和intein分裂系统。基于GFP突变衍生的EGFP和sfGFP由11个围绕中心α-螺旋的反平行β-折叠组成,可应用于分裂蛋白的研究[19, 22]。GFPN和GFPC依赖内在相互作用,非共价结合形成功能性GFP复合物,使得GFP分割位置有较大限制,严格依赖两端分裂肽的构象可塑性和亲和力。我们依据GFP蛋白二级结构中的11个β-片层结构,尝试了多种分割方案,GFPC9-11是其中最短的碳端分裂肽(数据未展示)。Zhao等[9]最近应用intein系统,获得IntN和IntC之间高亲和力连接EGFP分裂肽(N1-7/C8-11, aa 1~157/aa 158~238),显示了高效的拼接效率,且拼接后intein可自身移除。参考上述研究,本研究将intein分裂系统引入EGFPN1-8/C9-11,证实其能够显著增加EGFP分裂蛋白的拼接效率。进一步尝试应用intein策略分割EGFPN1-10/C11,但并未获成功,推测与EGFPN1-10构象可塑性较弱有关。因此,本研究尝试应用具有改良折叠构象的sfGFP,发现共表达sfGFPN1-10/C11分裂肽能够互补形成功能性荧光蛋白,但效率较低,与早先报道一致[19, 20, 23]。非常有意思的是,引入intein分裂系统显著增加了sfGFPN1-10/C11分裂肽的功能性拼接(图3B),推测sfGFPN1-10/C11分裂肽具有较好的构象可塑性,能够稳定表达,融合intein分裂肽则能显著增加它们之间的亲和力。
HBV 3.2 kb基因组高度压缩,每一个碱基都具有病毒蛋白编码功能以及可能的顺式调控功能,后者涉及HBV cccDNA转录调控、负链DNA合成中的蛋白引物转位以及rcDNA形成过程中正链DNA的延伸和转位等。另一方面,HBc富含精氨酸的碳端结构域精确调控病毒RNA基因组的包装,过长的病毒基因组则易造成核衣壳不稳定[24-25]。rHBV复制子通常依赖反向遗传学策略,删除已知的、非顺式调控区的部分序列,并插入大小合适的外源报告基因,删除的基因可通过反式互补而拯救重组病毒复制。本研究在HBV RNA加尾信号下游删除约339 bp的HBc蛋白编码序列,构建了HBV1.1-ΔHBc113载体。ΔHBc113载体实际上代表了一种HBc缺陷的病毒基因组,由于大片段缺失(339 bp),病毒Pol编码基因阅读框向5′端大幅前移,更加接近转录起始点。该载体在HBc互补条件下,能够在转染细胞中形成高效的病毒DNA复制(图1B)。在ΔHBc113载体中插入可用的报告基因则复杂得多,本研究首先在病毒Pol基因上游融合T2A编码序列,使得Pol与上游外源基因融合,通过断裂肽获得独立表达。尽管如此,大片段插入完整的荧光素酶或GFP序列使得ΔHBc113融合载体完全失去复制能力。有意思的是,当插入小片段(约<360 bp)GFP部分编码序列,Southern blotting显示明显的单链DNA(single-stranded DNA, ssRNA)形成(数据未展示)。这些结果提示,外源基因容量是rHBV细胞内复制的重要限制因素。
本研究利用intein/GFP分裂系统寻求功能性的GFP分裂短肽,以构建可视化的rHBV复制子。结果表明,ΔHBc113/sfGFPC11携带的外源序列仅编码IntC(39 aa)融合的sfGFPC11(16 aa)以及下游的T2A(21 aa),在HBc互补条件下能支持细胞内重组病毒的功能性复制(ssRNA + rcDNA),并产生子代病毒粒子(图4)。应该指出的是,ΔHBc113/sfGFPC11荧光强度弱于野生病毒复制子,故仍有较大的改进空间。综上所述,本研究设计构建了一种GFPC分裂蛋白融合的rHBV复制子,能够在共表达HBc以及GFPN的细胞中建立病毒复制,并产生功能性荧光蛋白。未来的工作将基于ΔHBc113/sfGFPC11子代病毒粒子从头感染sfGFPN1-10表达细胞,建立HBV可视化细胞培养模型,期望可用于进一步深入研究HBV复制机制以及抗病毒药物筛选。
