凋亡相关斑点样蛋白敲除PK-15细胞系的建立及对PRV感染的影响
2021-02-27翟云云李佳佳任子昱金前跃杜永坤
翟云云,李佳佳,张 爽,任子昱,金前跃,杜永坤*,万 博*
(1.河南农业大学动物免疫学国际联合研究中心,郑州 450002;2.河南省动物免疫学重点实验室,郑州 450002)
猪伪狂犬病(porcine pseudorabies, PR)是由伪狂犬病病毒(pseudorabies virus, PRV)引起的以仔猪神经系统紊乱和高死亡率、育肥猪呼吸障碍和生长缓慢以及母猪繁殖障碍等为主要特征的病毒性传染病[1]。PRV属于疱疹病毒科,为双链DNA病毒。先天免疫作为宿主防御病原体入侵的第一道防线,通过激活宿主的模式识别受体(PRR),识别大批病原体中保守的分子结构,即所谓的病原体相关分子模式(PAMP)。识别后,PRR被触发,激活Ⅰ型干扰素(IFN)、IFN刺激基因(ISG)和炎性细胞因子,从而抑制病原体的复制并促进适应性免疫反应[2]。同时病毒也有一套逃逸宿主天然免疫的机制,有研究表明,PRV在感染初期可以抑制大多数具有有效抗病毒作用的IFN-β刺激基因的表达[3-4],以便于逃避天然免疫。但是自2011年以后,以HeN1株为代表的PRV变异株在全国多个地区暴发流行,原有疫苗对猪群保护效率不足[5],因此伪狂犬病已经成为当前我国养猪业健康发展的重要威胁[6]。
凋亡相关斑点样蛋白ASC(apoptosis-associated speck-like protein containing a CARD)含热蛋白结构域(pyrin domain, PYD)和半胱天冬酶募集域(caspase recruitment domain,CARD),2个结构域均相对保守,都含有疏水性氨基酸,且都含有6个保守的α-螺旋,可以实现自身的寡聚化,并招募一些含有同源结构域的蛋白,共同参与炎症反应[7]。ASC参与多种炎症小体的形成,如NLR蛋白或AIM2等,可作为共有的接头蛋白与半胱天冬酶-1(caspase-1)共同激活下游炎症因子的产生[8]。
近年来,对ASC在病毒感染过程中引发的天然免疫方面的作用有越来越多的研究。黏液瘤病毒(myxoma virus)编码的M13L蛋白可与ASC互作,抑制caspase-1的激活,进而阻止炎症反应的发生[9]。HSV-1和腺病毒DNA感染通过激活典型炎症小体NLRP3、ASC和caspase-1通路导致IL-1β、IL-18的产生[10-11]。活化的ASC与STING相互作用并阻止TBK1和STING之间的缔合,从而抑制了Ⅰ型IFN的诱导[12]。dsDNA病毒在猪细胞系中以STING依赖性方式激活IFN-β[13],促进IFN-β的表达来抵御病毒的入侵。由于ASC与STING的相互作用从而抑制了具有抗病毒作用的IFN-β的表达,而ASC对PRV感染过程中IFN-β的表达是否存在调节作用还未见报道。为了更为全面地了解ASC在抗病毒天然免疫的作用,以及是否参与PRV逃逸宿主天然免疫的过程,首先通过CRISPR/Cas9技术构建猪ASC基因敲除PK-15胞,利用PRV-GFP模式病毒和PRV-QXX毒株检测ASC基因敲除对PRV增殖的调节作用,以期为进一步研究伪狂犬病病毒逃逸宿主免疫应答机制提供新思路,同时,为猪伪狂犬病的防治提供新的研究策略。
1 材料与方法
1.1 主要试剂材料
PK-15(猪肾上皮细胞)、Vero(非洲绿猴肾上皮细胞)和HEK293T/17(人胚肾上皮细胞衍生细胞系)购自美国ATCC;猪伪狂犬病病毒(PRV-QXX)为本实验室保存。猪伪狂犬病重组荧光病毒(PRV-GFP)由中国科学院武汉病毒所馈赠;E.coliTop10感受态细胞购自北京鼎国昌盛公司;LentiCRISPR v2(Addgene Plasmid 49535)、pMD2.G和pSPAX2购自美国Addgene公司;Q5高保真DNA聚合酶、限制性内切酶、T4 DNA连接酶和T7 E1酶购自美国NEB公司;Puromycin和细胞培养耗材均购自美国Thermo公司;反转录试剂盒、荧光定量PCR试剂、DNA Marker均购自大连宝生物公司。
1.2 引物设计与合成
参照美国国家生物技术中心(NCBI)数据库已公布的猪ASC基因组序列(GenBank登录号NC_010445.4)设计sgRNA和PCR扩增引物(表1);通过NCBI数据库查询需要检测的基因mRNA序列,通过Primer7.0软件设计Q-PCR引物(表1)。

表1 引物序列表
1.3 p-ASC-sgRNA载体构建
将两对sgRNA引物先进行退火杂交,退火条件95 ℃ 5 min。将LentiCRISPR v2质粒用BsmbⅠ酶37 ℃ 酶切3 h,将目的片段进行回收。将退火杂交的引物与酶切后回收的目的片段用T4 DNA连接酶在4 ℃条件下连接过夜。将连接产物转化至E.coliTop10感受态细胞,加入1 mL的无抗LB液体培养基,放入37 ℃摇床中,220 r·min-11 h。将培养后菌液均匀涂布在含有Amp的固体LB培养基中,37 ℃培养12 h。挑取单个菌落进行基因测序鉴定。用QIAGEN质粒中提试剂盒提取测序正确的质粒,分别命名为p-ASC-sgRNA1、p-ASC-sgRNA2。
1.4 ASC敲除细胞系的构建
利用慢病毒包装质粒(pMD2.G、pSPAX2)与构建成功的重组质粒共转染HEK293T/17细胞,于48 h回收慢病毒,并取1 mL病毒液感染PK-15细胞。感染48 h后,用含有8 μg·mL-1Puromycin的10% FBS/DMEM筛选细胞。待对照组细胞全部死亡时,将感染组换为3 μg·mL-1Puromycin的10% FBS/DMEM并将多克隆细胞进行扩大培养。取多克隆细胞提取基因组并进行T7酶切检测,通过有限稀释法将编辑效率高的多克隆细胞进行单克隆细胞系筛选。通过对测序结果进行比对,将敲除单克隆细胞株进行扩大培养用于后续试验。
1.5 T7核酸内切酶检测
以提取的基因组为模板,利用p-ASC-F/p-ASC-R PCR引物进行PCR扩增,琼脂糖凝胶电泳并回收PCR产物。取200 ng进行退火杂交,取0.6 μL T7核酸内切酶酶切1 h。用DNA PAGE胶电泳后用含有SYBR Green核酸染料的1×TBE溶液中染色1 h,通过在凝胶成像仪中对染色的DNA PAGE胶拍照来检测T7酶切结果,再利用ImageJ v1.8.0 2对编辑效果进行分析。
1.6 细胞增殖试剂盒(CCK-8)检测
以每孔1×104个细胞在96孔板中接种PK-15和PK15-ASC-/-细胞,各6孔。于不同时间点(0、6、12、24、36、48 h),在培养孔中加入10 μL CCK-8溶液37 ℃孵育2 h,450 nm波长检测培养液吸光度。
1.7 流式细胞仪检测
将PK-15和PK15-ASC-/-细胞以3×104个·孔-1接种于24孔细胞培养板,待细胞密度达到40%,空白细胞计数后以PRV-GFP MOI=0.01感染细胞,37 ℃培养箱中吸附 1 h后弃去原培养基,PBS清洗2遍后加入维持培养基后继续培养,分别于0、6、12、24、36、48 h进行流式检测分析。
1.8 Real-time quantitative PCR检测
以每孔3×105个细胞在35 mm培养皿中接种PK-15和PK15-ASC-/-细胞。以MOI=1 感染PRV-QXX后0、6、12、24、36、48 h收取RNA。Trizol裂解法提取细胞总RNA并通过去除染色质DNA、反转录成cDNA,以cDNA为模板,每个模板做3个重复孔,进行RT-qPCR检测。利用T-test对Q-PCR结果进行分析检验。
1.9 Western blot检测PRV-gE蛋白的表达
将PK-15和PK15-ASC-/-细胞按照6×105个接种于60 mm细胞培养皿中。待细胞密度达到60%,以MOI=1 PRV-QXX感染细胞。分别于0、6、12、24、36 h不同时间点后收获细胞,用90 μL RIPA裂解液(含蛋白酶抑制剂)裂解后用BCA方法测定蛋白浓度。取30 μg蛋白样品,进行SDS-PAGE凝胶电泳,然后通过湿转法将蛋白转移到PVDF膜上,用5%脱脂奶粉溶液室温封闭1 h。PRV-gE单克隆抗体(1∶1 000稀释)4 ℃过夜孵育,1×TBST以10、10、5、5 min的时间洗膜4次,然后室温孵育山羊抗鼠IgG二抗(1∶5 000)2 h。1× TBST以10、10、5、5 min的时间洗膜4次后进行显影,试验以β-actin作为参照基因。
1.10 PRV子代病毒滴度测定
以每孔3×105个细胞在35 mm培养皿中接种PK-15和PK15-ASC-/-细胞,每种细胞各种6个培养皿。培养细胞融合度至约40%,空白细胞计数后按照MOI=0.01 PRV-QXX感染细胞,37 ℃、5% CO2细胞培养箱中吸附1 h,吸附结束后,用PBS清洗1遍,换成维持培养基进行培养。按照0、6、12、24、36、48 h不同时间点收取样品,将病毒悬液反复冻融3次,离心取上清。以每孔1×104接种Vero细胞于96孔板,待细胞贴壁后,以10倍递次稀释将病毒液进行稀释,分别加入相对应的孔板中,37 ℃、5% CO2细胞培养箱中吸附1 h,结束后换成维持培养基进行培养。培养5~6 d,在显微镜下对病变孔进行统计,然后利用Reed-Muench法计算PRV子代病毒滴度。
1.11 统计学分析
各试验均平行重复3次,通过GraphPad 8.0软件将3次试验结果进行统计学分析并作图。
2 结 果
2.1 Cas9/sgRNA载体的构建
用BsmbⅠ对LentiCRISPR v2载体单酶切。37 ℃酶切3 h后进行琼脂糖凝胶电泳。电泳结果显示,在13 000和1 873 bp位置出现两个特异性条带,通过DNA胶回收试剂盒对13 000 bp目的片段进行胶回收后与sgRNA引物退火产物进行连接,通过转化将连接产物转入E.coliTop10感受态细胞。挑取单个菌落进行DNA测序。测序结果表明,重组质粒p-ASC-sgRNA构建成功(图1A、B),提取质粒用于后续试验。
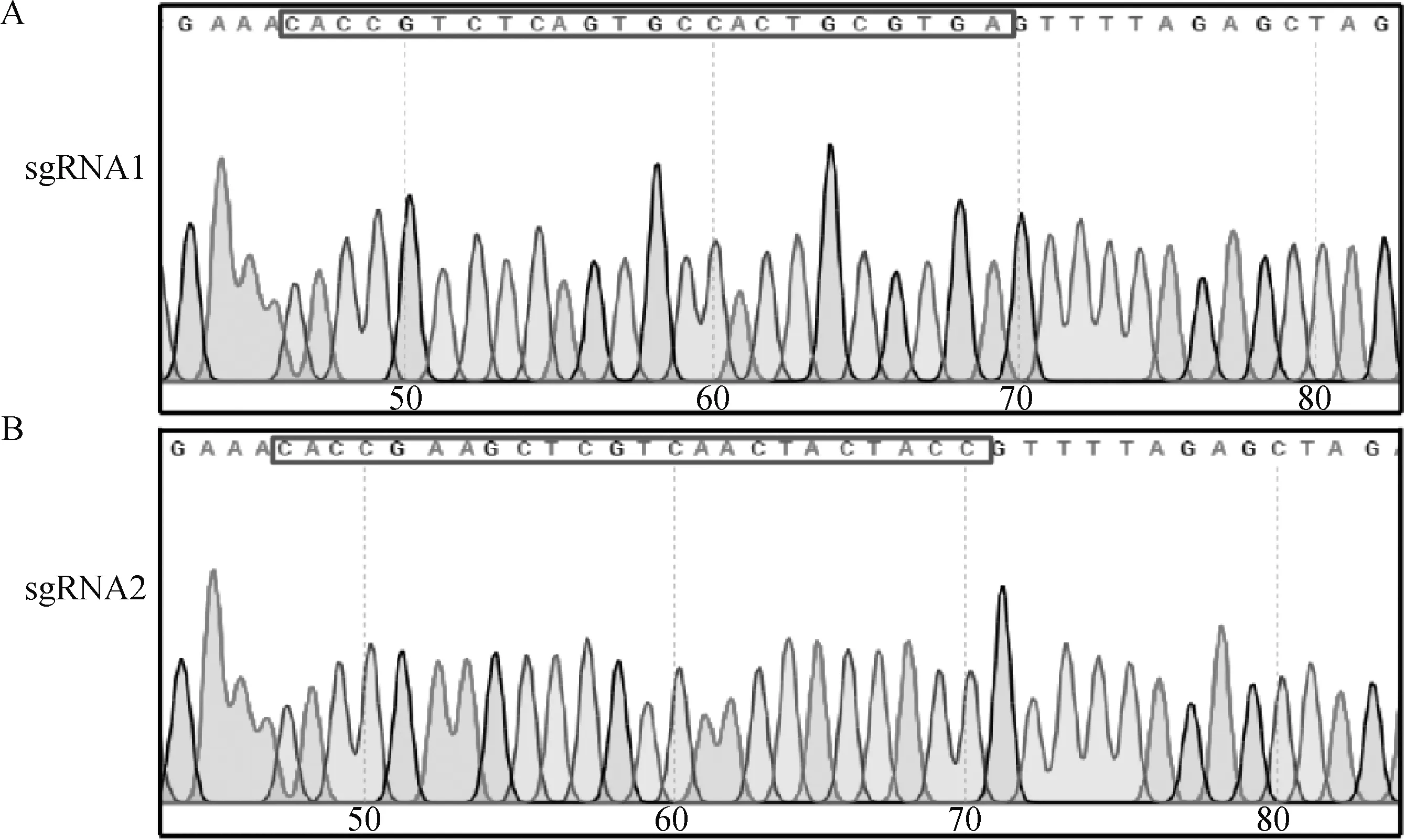
A.ASC-sgRNA1测序结果;B.ASC-sgRNA2测序结果;框内为sgRNA序列
2.2 PK-15敲除ASC基因单克隆细胞系的构建
T7酶切检测显示sgRNA1、sgRNA2均能有效对ASC基因进行编辑(图2A、B)。选取编辑效率较高的ASC-sgRNA2多克隆细胞进一步筛选PK15-ASC-/-单克隆细胞株,最终测序比对得到靶位点处插入了1个T碱基,并引起ASC后续密码子移码突变的8号单克隆细胞(图2C),并命名为PK15-ASC-/-。
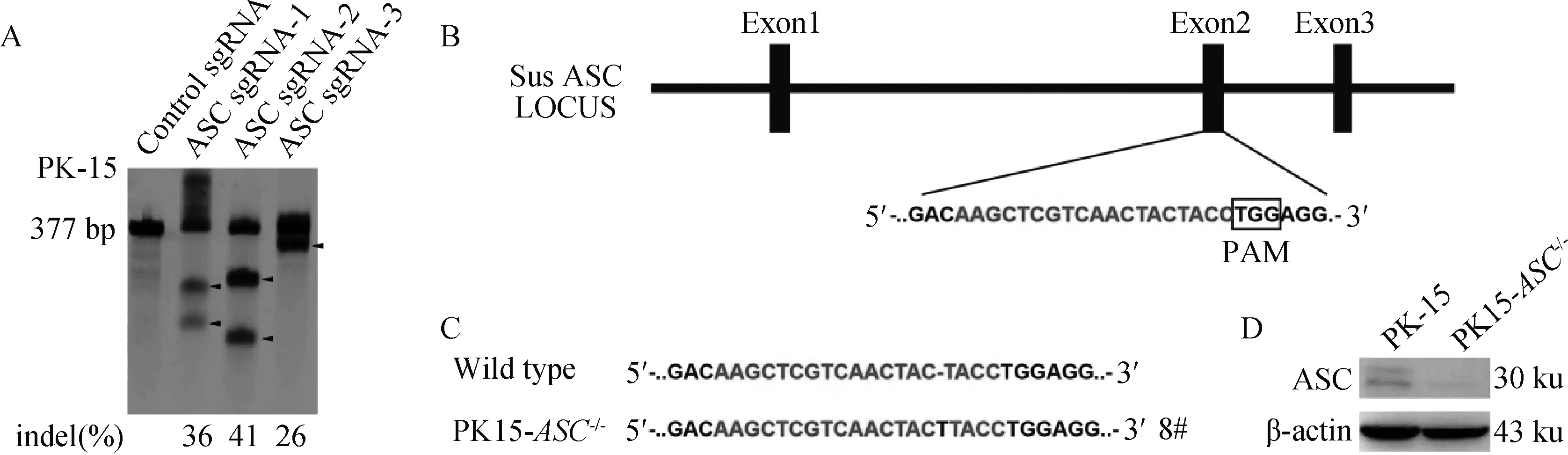
A.T7酶切鉴定ASC-sgRNA编辑效率(indel.Insert、Dellet、Mutation的缩写);B、C.敲除ASC基因的单克隆细胞系测序结果;D.Western blot鉴定ASC基因敲除的单克隆细胞
2.3 ASC基因敲除对PK-15细胞增殖的影响
通过CCK-8检测敲除ASC基因后,是否会影响细胞增殖活性。结果表明,敲除ASC基因后PK15-ASC-/-细胞与对照组相比OD450 nm波长吸光度值差异不显著,结果表明,ASC基因的敲除对PK15细胞的增殖无影响(图3)。
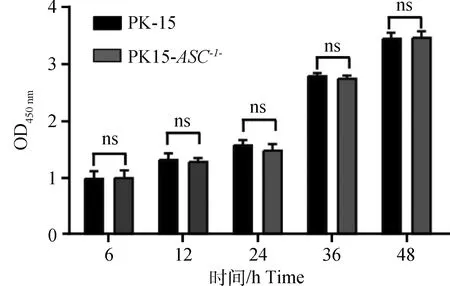
ns.P>0.05
2.4 敲除ASC基因在PK-15细胞中对PRV-GFP增殖的影响
用PRV-GFP(MOI=1)感染PK-15和PK15-ASC-/-细胞,分别在0、6、12、24、36、48 h后利用流式细胞仪检测细胞中GFP荧光强度。结果表明PK-15细胞中GFP荧光强度随着时间的增加显著升高,而PK15-ASC-/-细胞中GFP荧光强度与PK-15细胞中GFP荧光强度相比则显著减弱,表明敲除ASC基因后显著抑制PRV-GFP的增殖(图4)。
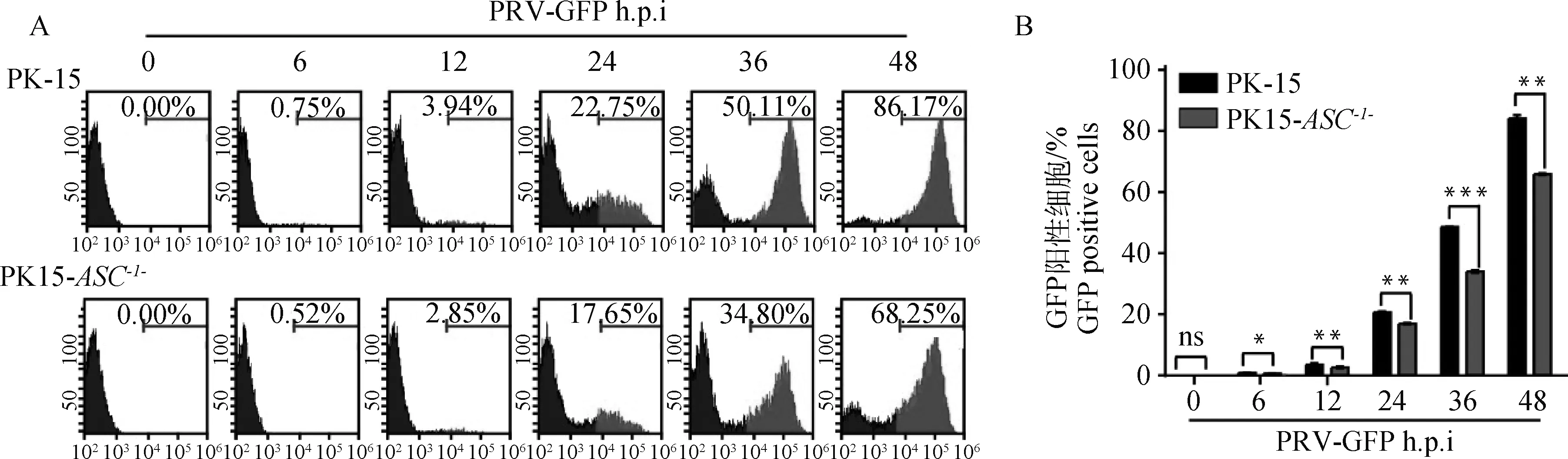
A.PRV-GFP流式检测结果;B.流式结果统计分析(ns.P>0.05;*.P<0.05;**.P<0.001;***.P<0.000 1)
2.5 ASC基因敲除对PRV-gB mRNA表达的影响
按MOI=1 PRV-QXX感染PK-15和PK15-ASC-/-细胞,RT-qPCR检测不同时间点PRV-gBmRNA表达量。随着PRV复制,PRV-gBmRNA表达量递增。敲除ASC后PRV-gBmRNA水平显著低于对照组(图5),结果进一步表明PK15-ASC-/-细胞相比于PK-15细胞可以显著抑制PRV基因转录。
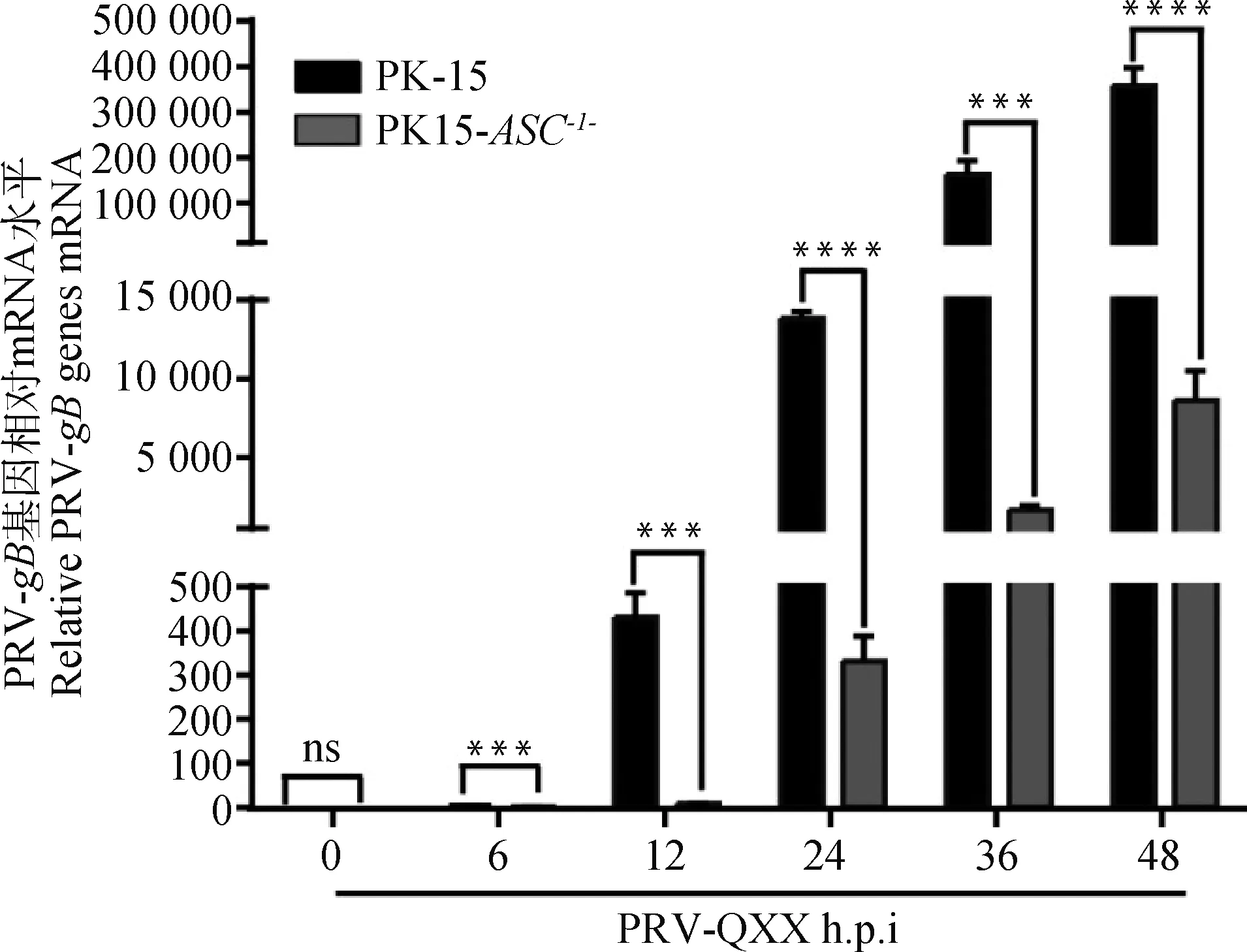
RT-qPCR检测感染PRV-QXX不同时间PRV-gB基因拷贝数;ns.P>0.05;***.P<0.000 1;****.P<0.000 09
2.6 ASC基因敲除对IFN-β、ISG15和IL-1β mRNA水平的影响
RT-qPCR检测PK-15和PK15-ASC-/-细胞感染PRV-QXX后细胞内IFN-β、ISG15 和IL-1βmRNA表达水平。如图6所示,PK-15与PK15-ASC-/-细胞内IFN-β、ISG15 mRNA表达水平上调,并与PRV感染时间呈现正相关,同时IL-1βmRNA的表达水平下调。且PK15-ASC-/-细胞与PK-15细胞相比组间差异显著。结果显示,敲除ASC基因抑制了IL-1βmRNA的表达,促进了IFN-βmRNA的表达,从而促进干扰素刺激因子的生成,抑制病毒感染。

PRV-QXX感染不同时间点RT-qPCR检测基因拷贝数;A.IFN-β;B.ISG15;C.IL-1β;ns.P>0.05;*.P<0.05;***.P<0.000 1;****.P<0.000 09
2.7 ASC基因敲除对PRV-gE蛋白表达的影响
Western blot检测结果(图7)显示PK-15细胞感染PRV 24 h后可以检测到PRV-gE蛋白,36 h后表达量最高。虽然PK15-ASC-/-细胞在24 h也可以检测到PRV-gE蛋白,但在24、36 h的组间灰度分析结果显示PK15-ASC-/-细胞中PRV-gE蛋白表达显著低于PK-15细胞。结果表明,PK15-ASC-/-细胞相比于PK-15细胞显著抑制PRV-gE蛋白的翻译。
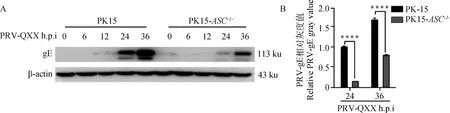
A.Western blot结果;B.灰度值分析结果;****.P<0.000 09
2.8 ASC基因敲除对PRV-QXX子代病毒滴度的影响
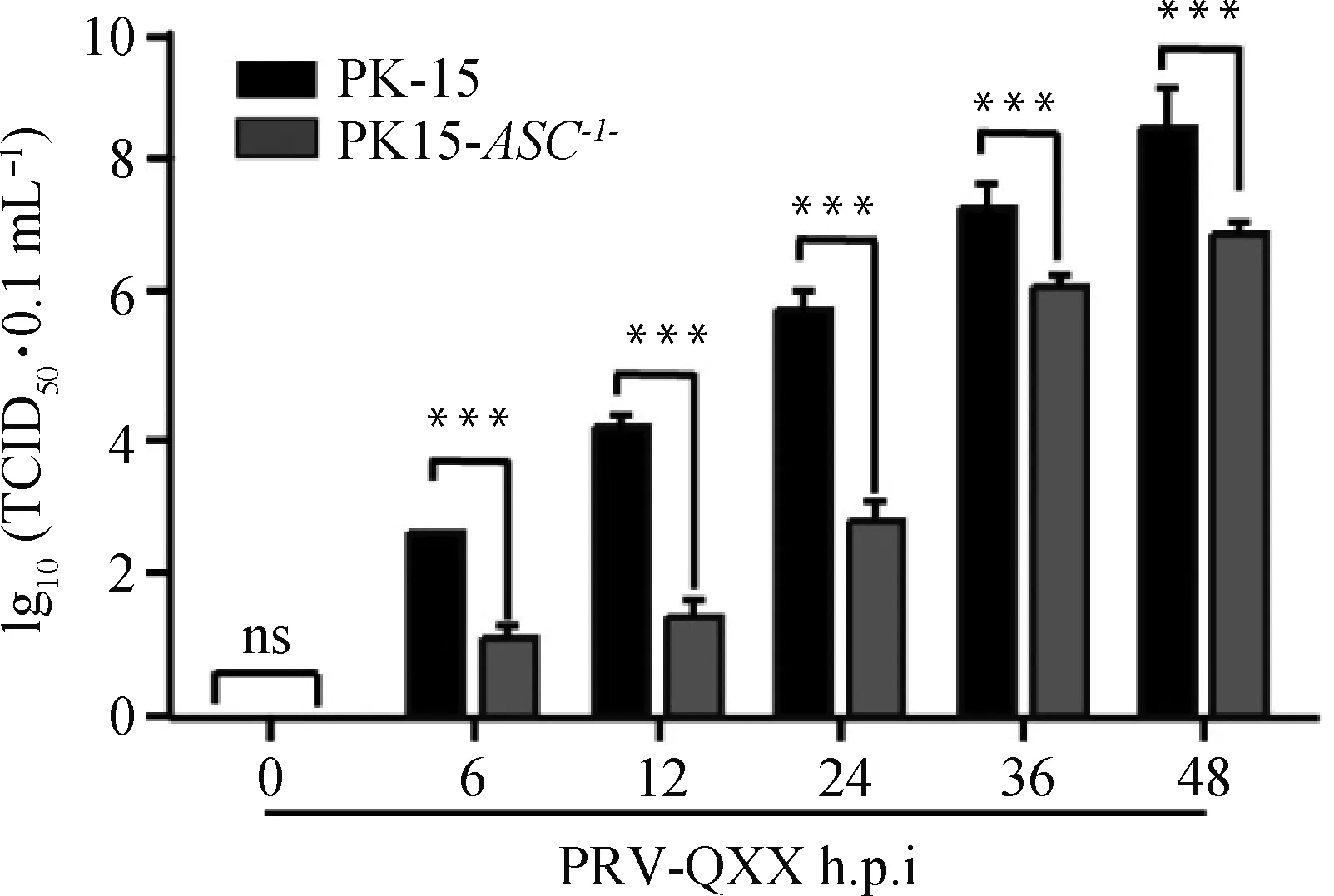
用PRV-QXX(MOI=1)感染PK-15和PK15-ASC-/-细胞,分别在0、6、12、24、36、48 h后收取子代病毒,利用TCID50法进行病毒滴度测定。结果表明PK15-ASC-/-细胞中子代病毒滴度与PK-15细胞中子代病毒滴度相比显著减弱,表明敲除ASC基因抑制PRV-QXX子代病毒的产生。
3 讨 论
ASC具有PYD和CARD 2个结构域,可分别结合NLRP3和caspase-1,形成炎性小体。ASC作为接头蛋白连接pro-caspase-1,使pro-caspase-1自分裂、活化,促进IL-1β和IL-18的分化成熟,同时诱导黏附分子、趋化因子等炎症细胞因子的合成,放大局部炎症反应[14]。有研究表明,ASC参与多种宿主抗病毒天然免疫过程。流感病毒的NS1蛋白与NLRP3相互作用并抑制了ASC诱导的炎性小体完全活化所需的单斑点形成,从而抑制NLRP3炎性小体介导的IL-1β分泌[15];ASC能够协调炎症小体依赖性和非依赖性免疫反应,以有效控制西尼罗河病毒(WNV)感染[16];ASC和STING之间的相互作用可以抑制STING及其下游分子TBK1的激活,最终抑制IFN-β的表达[12];而在缺乏ASC的巨噬细胞中poly(dA-dT)可以引起较高水平的IFN-β的表达[17]。
*.P<0.05; ***.P<0.000 1
PRV是一种重要的动物病原,其感染过程复杂,可以在靶细胞核内完成基因组装,然后通过出芽在细胞核膜内侧达到最终的成熟,进入细胞质。同时,病毒组装可以通过二次出芽进入高尔基体网的囊泡中形成最终的病毒粒子,通过囊泡质膜的融合最终完成成熟的PRV病毒粒子的释放。干扰素(IFN)是宿主先天性免疫系统是针对病毒感染的主要防御策略[18-20]。在PRV感染的早期阶段宿主细胞ISG15被上调,而体外试验中证实过表达ISG15增加IFN-β的表达,降低PRV的病毒滴度和mRNA水平,从而有效地抑制PRV复制[21]。外核苷酸焦磷酸酶磷酸二酯酶1(ENPP1)通过水解cGAMP,抑制IRF3磷酸化,减少IFN-β的分泌。过表达ENPP1抑制IFN-β表达从而增强PRV感染,而ENPP1的沉默使PRV感染减弱[22]。PRV感染,内化的病毒DNA以STING依赖性方式激活IFN-β,激活宿主体内的天然免疫抵抗病毒感染[13]。笔者的研究结果显示,与正常PK-15细胞相比,感染PRV后PK15-ASC-/-细胞IL-1β的表达显著降低,同时IFN-β的表达显著升高,试验首次证实ASC对PRV感染有显著调节作用。有研究报道PRV通过STING信号通路激活宿主体内的IFNs刺激因子的大量表达[13],而ASC可以与STING互作抑制其信号通路下游分子IFNs的表达[12],但是ASC是否会通过调节STING从而影响IFN-β的表达,需要更为深入的探究。同时,有研究表明,IFNs天然免疫与炎性小体信号通路间存在拮抗效应,Ⅰ型干扰素抑制炎性小体活性和IL-1β的产生[23-24],而炎性小体信号通路也可以负调控IFNs天然免疫信号通路激活,各种NLR(如NLRX1[25]、NLRP4[26]、NLRP6[27])对IFNs有负调控作用。本试验的研究结果也证实了敲除ASC抑制炎性小体信号通路同时能够激活IFNs天然免疫信号传导。试验结果表明,抑制炎性小体信号通路可以更好地抑制PRV的感染,但ASC在IFNs天然免疫信号通路具体的作用有待进一步研究。
本次试验利用CRISPR/Cas9基因编辑技术对猪ASC基因进行成功敲除后,研究ASC对PRV感染的影响。本研究选择递送效率更高的慢病毒载体系统,将Cas9和sgRNA导入到PK-15细胞系中,能大大提高ASC基因编辑效率。PRV体外感染试验结果表明敲除ASC基因能够显著抑制PRV感染。试验通过多种方法证实敲除ASC基因能够显著抑制PRV感染,首先通过流式细胞仪技术检测表明PK15-ASC-/-细胞中GFP荧光强度显著低于PK-15细胞;通过TCID50测定表明PK15-ASC-/-细胞中子代病毒滴度显著低于PK-15细胞;通过Western blot检测表明PK15-ASC-/-细胞中gE蛋白表达显著低于PK-15细胞;通过Q-PCR检测表明PK15-ASC-/-细胞中PRV-gBmRNA表达显著低于PK-15细胞。进一步机制研究发现,敲除ASC基因能够显著促进IFN-βmRNA和ISG15 mRNA表达。大多数ISGs表达后能够作用于病毒的整个生活周期(吸附、进入、组装和释放),抑制病毒进一步感染。但ASC在PRV生活周期发挥调节作用的具体阶段还需要进一步详细研究。
本试验为PRV致病机制的研究提供了新的思路,为PRV防控策略的制定提供了新的数据支撑。
4 结 论
利用慢病毒介导的CRISPR/Cas9基因编辑技术获得PK15-ASC-/-单克隆细胞系,通过流式细胞仪、病毒滴度测定、Western blot和RT-qPCR检测了PRV感染情况,结果表明,敲除ASC基因促进IFN-βmRNA的表达,从而抑制PRV感染。
