基于过渡金属二硫族化物析氢催化的研究进展
2021-02-26何倩倩孟令佳宫勇吉
何倩倩,王 哲,孟令佳,陈 乾,宫勇吉
(北京航空航天大学材料科学与工程学院,北京100191)
环境问题是全世界特别是工业化和发展中国家严重的公共卫生问题[1~5],据美国能源情报署(United States Energy Information Administration)预计,2012年至2040年,全球对于能源的年消费量将以每年48%的速度增长[2].所以必须开发不依赖化石燃料且具有可持续性的替代清洁能源[5].
氢能源(H2)是传统化石燃料的有效替代选项之一.H2具有高达120~142 kJ/kg的能量密度[1],同时H2是理想的“零污染”能源(H2+0.5O2→H2O).当前工业上主要采用天然气重整、煤气化和电解水3种方法制氢[图1(A)][3].电解水是绿色产氢的基本方法之一[6,7].实际工业中天然气重整和煤气化产氢占总产氢的95%.而进一步降低电化学产氢的成本是其推广的重要前提.

Fig.1 Three main pathways for industrial hydrogen production(A)and schematic diagram of an electrolyzer(B)[3]
自1789年电解水现象被发现以来,电解水产氢就引起了广泛的关注[8,9].电解水装置如图1(B)所示,分为两个半反应,产生氢气的半反应称为析氢反应(Hydrogen evolution reaction,HER)[3].通常电解水均需要催化剂来降低电化学过电位(电化学反应的施加电位和热力学电位之差),以提高能量转换效率,从而能够在较低的过电位下获得较高的电流密度[10,11].目前,最有效的HER催化剂是铂族金属[12],然而,由于贵金属的稀缺产量和昂贵价格阻碍了HER的发展.
近年来出现了一类新型催化剂——过渡金属二硫族化物(Transition metal dichalcogenides,TMDs),相比贵金属而言,TMDs产量大、价格低、催化活性好、便于调控和修饰[13,14].本综述介绍了HER的原理,列举了近10年来具有代表性的一些研究工作,讨论了原子工程(硫空位、结构缺陷、掺杂合金、插层)、相工程、异质结对TMDs催化性能的调控(如图2和表1所示),总结和展望了TMDs的研究进展以及该领域面临的挑战和机遇.
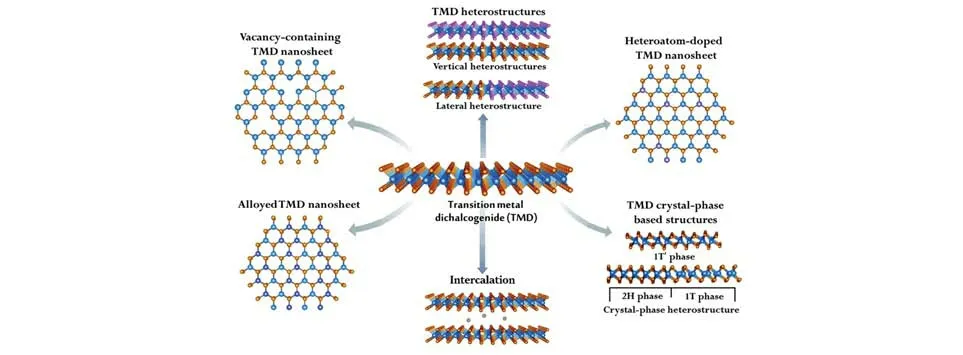
Fig.2 Schematic illustration of the methods to tune the catalytic properties of TMDs nanosheets[13]
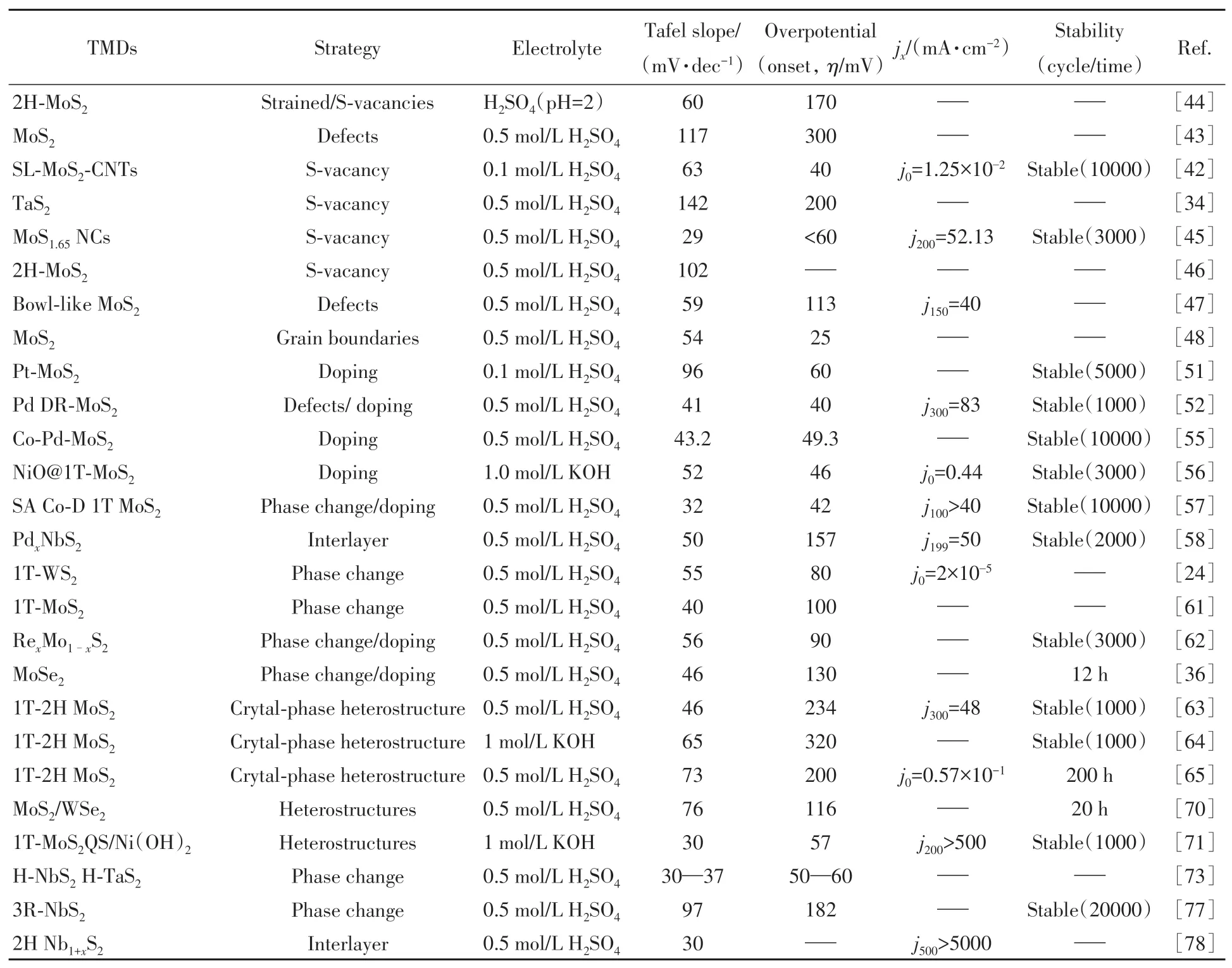
Table 1 Summary of hydrogen evolution catalytic performance of some TMDs under different control methods
1 氢气析出反应的电极反应原理
出于对能量消耗的考虑,电解水一般在质子交换膜的酸性环境中进行,总的反应为

水分解反应包括2个半反应:
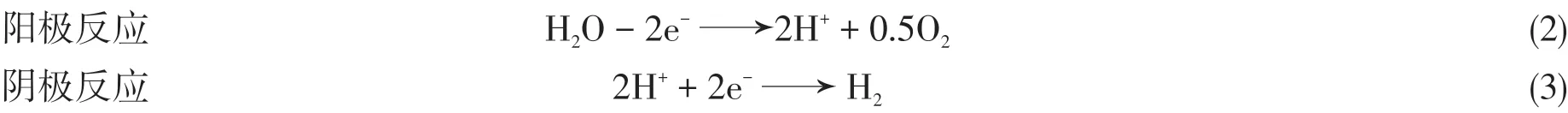
酸性溶液中,阴极的HER过程包括3个基元反应[3,14~16]:(1)Volmer反应一个质子和一个电子结合,在电极表面产生一个吸附态的氢原子(2)Tafel反应两个吸附态的结合产生氢气;(3)Heyrovsky反应2吸附态的吸收一个电子,同时结合溶液中的一个质子,从而产生氢气.
2 TMDs的HER催化性能
TMDs的化学式通式为MX2(M为Mo和W等过渡金属,X为硫族元素如S和Se).单层TMDs中常见的相结构包含1H相(三棱柱形)和1T相(正八面体形)[13,17,18][图3(A)],2或3个1H相的单层堆叠可以形成2H相(六边形结构)和3R相(棱形结构)[13].1T′则可视为扭曲的1T相.不同的M具有不同的稳定相及不同的催化性能.理论研究表明,在无任何外力的作用下,对于Mo和W的TMDs而言,1T相是亚稳态的,而H相通常是热力学稳定的结构[17~21],通常,亚稳态的T相在HER的应用中表现出更好的性能,且单层和少层也表现出良好的性能[22~24].
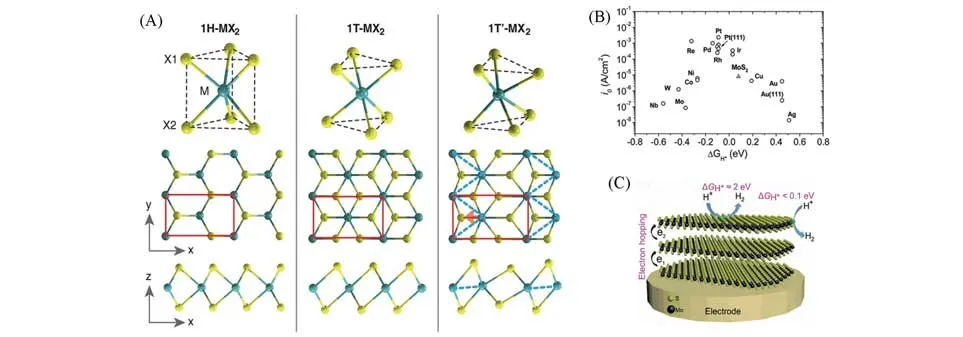
Fig.3 Schematics of 1H⁃MX2,1T⁃MX2,and T´⁃MX2(A)[18],volcano plot of the exchange current density as a function ofΔGH*for nanoparticulate MoS2 and the pure metals(B)[28],electron hopping in multilayered 2H⁃MoS2 and theΔGH*at basal plane and edge sites(C)[14]
氢吸附自由能ΔGH*可用于描述HER的活性,判断某个材料是否为良好的催化剂的标准是吸附氢的自由能与反应物或产物的自由能是否接近,即ΔGH*≈0[15,25~27].2005年,Hinnemann等[15]首次利用密度泛函理论(DFT)分析了MoS2边缘的氢原子吸附能,大约为0.08 eV[26],与Pt非常接近,位于“火山峰”附近[图3(B)],表明MoS2具有良好的催化活性,之后大量研究表明MoS2,WS2等TMDs具有边缘活性(ΔGH*<0.1 eV),而面内表现出惰性(ΔGH*≈2 eV)[图3(C)][14,28~31],随后很多研究表明,MoSe2,WSe2等TMDs的边缘也具有很高的HER催化活性[22,24,30~35].
通常,TMDs的HER催化性能和两个方面密切相关:(1)催化活性位点.边缘部位在大多数情况下是唯一具有催化活性的位置[28~32,35],因此如何增加边缘位点的数量或提高面内位点的催化活性是重要的研究方向.(2)材料的电导率.TMDs的层间以及电极之间的电子传导情况影响着整体的催化性能[图3(C)][12,14,36~41],进一步提高TMDs材料的导电性对其性能也有重要的影响.
对于TMDs限制因素的理解,为研究和提高TMDs催化性能指明了方向,可以基于以上两点来调控TMDs的催化性能.
3 TMDs的析氢性能调控
3.1 原子工程
3.1.1 硫空位 由于TMDs独特的层状结构,往往会存在如M原子空位和X原子空位等很多缺陷.这些缺陷主要为生长过程和后处理(如Plasma)产生的缺陷.研究表明这些缺陷有利于提高TMDs对HER的催化活性[42~50].
MoS2是 一 种 有 效 替 代 贵 金 属 的HER催 化 剂[28,29],通 常 减 少MoS2的 层 数 也 可 促 进HER的 活性[23,24,30,41,42],可能是减少层数时往往不可避免地引入缺陷.2014年,Deng等[42]利用水辅助法直接合成了不同层数的MoS2[图4(A)],在酸性环境中,以碳纳米管(CNT)为模板生产的单层MoS2(SL-MoS2-CNTs)表现出最佳的HER活性,研究表明,因为在减少层数时面内空位缺陷的引入[图4(B)],其起始电位相对于可逆氢电极仅为40 mV(vs.RHE),塔菲尔斜率为63 mV/dec[图4(C)].另外,通过后处理来产生缺陷也是研究热点之一.Li等[34]报道了用O2plasma处理TaS2纳米片[图4(D)].同年,Ye等[43]通过O2plasma和H2退火分别处理单层MoS2[图4(E)],O2plasma和氢气处理使得材料表面形成大量的裂纹和高密度微孔,暴露更多的活性边缘从而提高催化活性.随着Plasma的轰击,Tafel斜率由342 mV/dec变成171 mV/dec(Plasma处理20 s后)[图4(f1)和(f2)];氢气500℃下退火的MoS2较其它退火温度具有最低的起始过电位和最大的电流密度[图4(f3)和(f4)].

Fig.4 Schematic illustration for the direct chemical synthesis of different layer MoS2(A),HAADF⁃STEM image of defects in FL⁃MoS2 nanosheet(up)and HRTEM images of SL⁃MoS2⁃CNTs(down)(B),electrochemical measurement results of different layer MoS2(C)[42],schematic de⁃scription of plasma treatment of TMDs nanosheets(D)[34],morphology of pristine monolayer MoS2(e1)and monolayer MoS2 treated by O2 plasma(e2)and annealed by H2(e3)(E),electro⁃chemical measurement results of MoS2 before and after treatment(F)[43]
Li等[44]用Ar plasma处理MoS2,创新地提出在面内引入S空位和应变来激活2H-MoS2的面内活性,研究指出(1.35±0.15)%应变和(12.5±2.5)%S空位具有最佳的催化活性:ΔGH*≈0,Tafel斜率为60 mV/dec,过电位为170 mV(vs.RHE).研究表明,S空位和应变相结合的面内缺陷可以激活TMDs的面内活性,但是这两个因素会引起材料结构稳定性下降,在一定程度上限制了该方法的实际应用.
Lin等[45]用阳离子交换树脂来制备贫硫的单层二硫化钼(MoS1.65)[图5(A)],DFT计算表明,单层MoS1.65的表面和边缘都具有金属性[图5(B)],大量硫空位是造成表面金属性的原因.在酸性环境中,原始的单层MoS2的Tafel斜率为51 mV/dec,起始过电位为120~140 mV(vs.RHE),而优化的贫硫MoS1.65的Tafel斜率为29 mV/dec,优于Pt(34 mV/dec),起始过电位为60~75 mV(vs.RHE)[图5(C)],是迄今开发的最佳催化剂之一[34,42~47].2017年,Tsai等[46]使用电化学脱硫来优化2H-MoS2的面内活性,面内的S原子被氢化,以H2S的形式脱去[图5(D)],从而在面内形成S空位,一旦硫原子被电化学还原,则催化剂被永久活化.研究表明,当S空位浓度在12.5%~15.62%之间时ΔGH*=0[图5(E)],具有最佳的面内活性,实验结果显示,处理后的催化剂(V-MoS2)的电流密度明显高于原始(P-MoS2)材料,电流密度增加量与原始材料电流密度的比值ΔJ/J0=(438±159)%[图5(F)],且V-MoS2的Mo转化率(TOFMo)比P-MoS2的高了一个数量级,并且与文献报道的V-MoS2[44]相当[图5(G)].可见,S空位不仅优化了MoS2面内活性,还改善了MoS2与电极之间的电子传输,二者的协调作用促进了HER的催化活性.

Fig.5 Schematic showing the metallic edge,near⁃edge regions,and semiconducting core of the MoS2 NCs(A),calculated density of state(DOS)of MoS2 in the core and/or edge region,and the entire NC(left)and decomposition of the total DOS of MoS2 with S vacancies(S depletion)in the core and edge regions into partial DOS of the Mo and S orbitals(right)(B),polarization curves of dif⁃ferent catalysts in 0.5 mol/L H2SO4(C)[45],free energy diagram for the protonation and removal of S(D),hydrogen adsorption free energy onto a sulfur atom on the basal plane for each concen⁃tration of S⁃vacancies(E),polarization curves(F)and Tafel plots of monolayer MoS2(G)[46]
3.1.2 结构缺陷与晶界 最近,Zhou等[47]设计了一种新的MoS2缺陷结构[图6(A)和(B)],利用改良的逐步化学气相沉积法在倾斜的SiO2/Si基底上合成了三角形碗状的MoS2纳米片(Bowl-like MoS2):中心厚0.87 nm,四周边缘厚15.5 nm.测试表明Bowl-like MoS2具有一定的铁磁性,在H⊥下,铁磁Bowl-like MoS2,层间的转移效率提高[图6(C)和(D)],在酸性环境下,过电位为113 mV(vs.RHE),Tafel斜率为59 mV/dec[图6(E)和(F)],而且在恒定外电势为—150 mV时,在H⊥下,铁磁Bowl-like MoS2薄片可提供大约2倍的电流密度[图6(G)].铁磁Bowl-like MoS2薄片在HER反应中具有良好的稳定性,且这种磁性促进Bowl-like HER活性的方式不仅可用于MoS2材料,还可用于其它二维过渡金属硫族化合物材料.晶界(Grain boundaries,GBs),作为TMD材料中的一类重要缺陷[48~50],He等[48]提出Au量子点辅助的爬升驱动0D/2D相互作用的富晶界薄膜的生长机制[图6(H)和(I)],合成了晶界密度高达1012cm—2的TMD薄膜[图6(J)和(K)].测试采用四电极微电化电池,在酸性环境中,制备的MoS2纳米晶粒薄膜表现出优异的电催化HER性能,起始电位为—25 mV(vs.RHE),Tafel斜率为54 mV/dec[图6(L)和(M)].值得注意的是,该方法可以适用于其它TMD材料,如WS2,是一种合成晶圆尺寸的亚10 nm晶粒原子薄膜的通用方法.
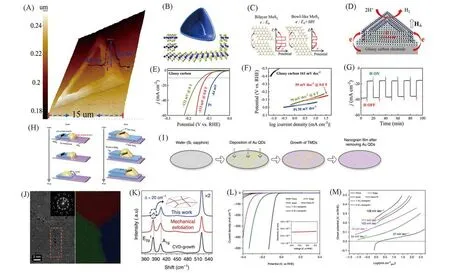
Fig.6 3D AFM image and cross⁃sectional step height profile of a bowl⁃like MoS2 flake(A),schematic dia⁃gram of a bowl⁃like MoS2 structure(B),schematic of electron transfer in bilayer MoS2 and bowl⁃like MoS2 under an external magnetic field(C),schematic of electron transfer in bowl⁃like MoS2 flakes during HER(D),polarization curves(E)and Tafel plots of different situation in 0.5 mol/L H2SO4(F),chronoamperometric responses(j⁃t)recorded from bowl⁃like MoS2 flakes at a constant overpotential of−150 mV(vs.RHE)under pulsed turn⁃on and turn⁃off magnetic field(G)[47],schematic of the climb stage and schematic of the drive stage(H),schematic of the wafer⁃scale growth of TMD nanograin films(I),HAADF STEM investigation of MoS2 grains(J),Raman spectra acquired from the MoS2 film(K),polarization curves(L)and Tafel plots of MoS2 devices in 0.5 mol/L H2SO4(M)[48]
3.1.3 掺杂合金 缺陷工程是激活TMD材料面内活性的有效方法,但研究发现,通过杂原子修饰也可触发惰性TMDs基面的HER催化性能.早在2015年,Deng等[51]首次证明了通过单原子金属取代MoS2中的“Mo”原子来触发惰性基面的催化活性[图7(A)],DFT计算表明,掺杂的金属原子可以调节氢原子在与金属原子相邻的S原子上的吸附行为[图7(B)],实验结果表明,M-MoS2面内S的ΔGH*≈0 eV,表现出优异的催化活性[图7(C)].他们还给出了多种金属元素(V,Zn)掺杂MoS2的H吸附能火山图[图7(D)].Qi等[52]通过理论和实验表明,富缺陷MoS2表面吸附Pd原子可有效调节MoS2表面电子态[图7(E)],Pd ND/DR-MoS2表现类似Pt的优异活性:低起始过电势[40 mV(vs.RHE)]和小Tafel斜率(41 mV/dec)[图7(F)和(G)].2019年,Pan等[53]采用第一性原理计算研究了过渡金属(TM)掺杂激活1T′-ReS2的面内催化活性,结果表明,用TM掺杂不仅可以通过调节氢原子在ReS2表面上的吸附行为来显著降低ReS2的ΔGH*,而且还可以暴露更多的活性位点,研究表明,用非贵金属Mo和Cr掺杂的ΔGH*接近于零[图7(H)],与贵金属Pt掺杂的ReS2相当.而且Hwang等[54]也使用DFT计算研究了48种不同TM-TMD组合的单原子催化剂,来鉴定多功能催化剂[图7(I~K)],研究表明,Pt-MoS2,Pd-NbS2和Pt-NbS2可作为特殊的双功能催化剂,还可以指导以极少含量的贵金属来开发高性能的催化剂.
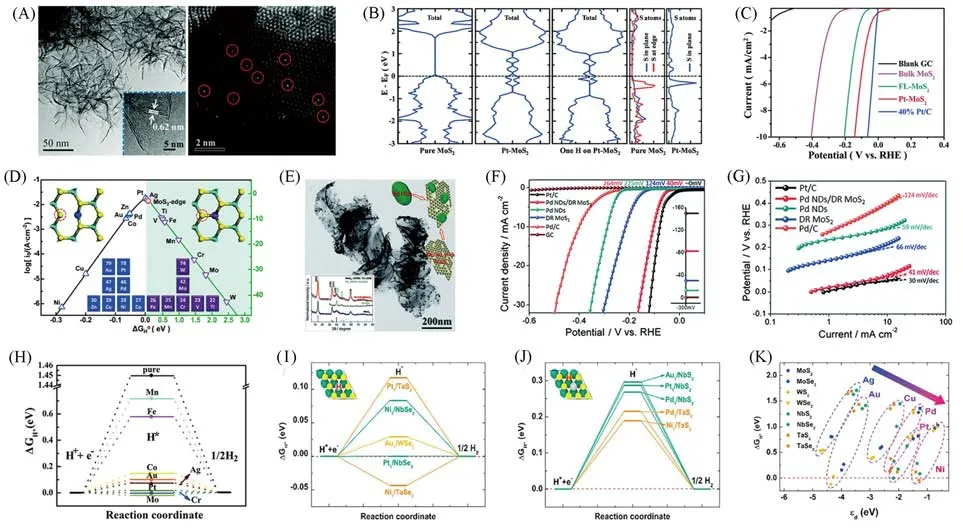
Fig.7 TEM image and HAADF⁃STEM images of Pt⁃MoS2(A),total DOS and projected DOS of pure MoS2 and Pt⁃MoS2,respectively(B),HER polarization curves of Pt⁃MoS2(C),volcano curve of different atom doped(D)[51],TEM image of Pd ND/DR⁃MoS2(E),polarization curves(F)and Tafel plots(G)of Pd ND/DR MoS2[52],ΔGH*vs.the reaction coordinate of HER for TM doped ReS2(H)[53],ΔGH*diagram of H adsorption on single atom(I)and on ligand next to single atom(J)and relations between the ΔGH*on single atom as a function of d⁃band center of single atom bound on a support(K)[54](A—D)Copyright 2015,Royal Society of Chemistry;(E—G)Copyright 2016,Royal Society of Chemistry;(H)Copyright
最近,Yang等[55]设计了一种原子嵌入辅助掺杂策略[图8(A)],实现了高活性、长寿命的双金属掺杂MoS2基HER催化剂.以MoO3为前驱体,将双金属Co,Pd均匀地插入到超薄MoO3纳米带的范德华(vdW)间隙中,然后通过硫化工艺获得了Co/Pd共掺杂的MoS2复合材料(Co-Pd-MoS2).这种策略的优点是可将各种杂原子诱导到MoO3的vdW间隙中,并将其牢固地锚定在vdW间隙之间,从而抑制了掺杂原子的聚集.酸性环境中,Co-Pd-MoS2的电化学性能接近5%Pt/C,远远超过纯的、单金属掺杂的催化剂,过电位为49.3 mV(vs.RHE),Tafel斜率为43.2 mV/dec,并且在长时间测试和循环过程中表现出优越的稳定性[图8(B)].DFT计算表明,晶格中的金属杂原子应该是对提高HER性能起着主导作用,通过自旋极化和Jahn-Teller(JT)效应,将Co和Pd原子诱导扭曲到硫原子上,导致其ΔGH接近于零,从而Co-Pd-MoS2表现出较高活性[图8(C)].

Fig.8 Schematic illustration of the synthesis of M⁃MoS2(A),electrochemical evaluation of HER perfor⁃mance for pristine,doped MoS2 and performance comparison in 0.5 mol/L H2SO4(B),Co⁃Pd⁃MoS2 with different local symmetries andΔGH diagrams of different catalytic sites of most stable doped structures in Co⁃Pd⁃MoS2(C)[55]

Fig.9 Structure characterizations of NiO@1T⁃MoS2:SEM,TEM,EDX mappings,HAADF⁃STEM and intensity profiles(green:Mo;orange:Ni)(A),polarization curves(B)and Tafel plots(C),stability tests(D)and electrochemical impedance spectroscopy(E)of NiO@1T⁃MoS2,first⁃principles calculations of the doping effect on HER performance(F,G)[56],schematic illustration of synthetic method for SA Co⁃D 1T MoS2 and its characterization(H),HER performance of SA Co⁃D 1T MoS2(I—N)[57]
贵金属是性能最好的催化剂,但是开发非贵金属也是科学家所关注的.2019年,Huang等[56]提出一种催化剂的设计策略,可同时增加活性位点的数量和每个现有活性位点的固有活性.他们将TM和O原子共掺杂到1T-MoS2的基面上[图9(A)],可有效地促进初始水离解和最终氢生成的动力学.碱性环境下,NiO@1T-MOS2起始电位接近于0 mV,过电位为46 mV(vs.RHE),Tafel斜率为52 mV/dec,并且具有优良的稳定性[图9(B~D)],且在200 mV下表现出最佳的电荷转移电阻[图9(E)].DFT显示,1T-MoS2中掺入TM会使ΔGH*降低,NiO@1T-MOS2的ΔGH*接近于0 eV[图9(F)和(G)].最近,Qi等[57]报道了一种简单的自上而下的组装策略[图9(H)],设计了一种独特的单原子阵列催化剂SA Co-D 1T MoS2(Co的质量分数为3.54%),面内引入Co单原子,其与S的协同作用有效提高了HER催化性能,酸性环境下,过电位为42 mV(vs.RHE),Tafel斜率为32 mV/dec,并且具有出色的长期循环稳定性,在100 mV(vs.RHE)过电位下显示出7.82 s—1的TOF[图9(I~N)],超过了文献[46]报道的非Pt基材料.进一步表明该方法具有普适性,可以扩展到任何符合标准的2D金属或金属氧化物.
3.1.4 插层 插层作为一种特殊的原子工程[57~60],当TMDs作为基体材料插入客体分子或者原子时,可改变层与层之间的相互作用,稳定晶体结构,扩大层间距,从而促进HER催化性能.Huang等[58]将Pd原子掺入NbS2的范德华层间来构造新的化合物PdxNbS2(x=0~0.23)[图10(A)和(B)],表现最佳的Pd0.23NbS2具有低的Tafel斜率(50 mV/dec)和过电位157 mV(vs.RHE),循环测试12 h内性能变化可忽略不计[图10(D~F)].理论计算表明,优异的性能源自Pd的原子柱效应,不仅扩展了NdS2的层间距,而且形成的PdS6八面体结构稳定了晶体结构[图10(C)],还以ΔGH*=0.06 eV的值优化了Pd周围的基础平面上S原子的催化活性[图10(G)和(H)].目前,将插层应用于电催化领域还比较少,但原子支柱可进一步应用于TMDs来设计新的催化剂.

Fig.10 Schematic of electron transfer in PdxNbS2 during HER(A),HAADF⁃STEM image of Pd0.23NbS2 along(B),schematic illustration of atomic⁃pillar effect of Pd in PdxNbS2(C),polarization curves(D),Tafel plots(E)and time⁃dependent potential curves(F)of Pd0.23NbS2,partial charge density of NbS2 and PdxNbS2 single layers within 1.0 eV below Fermi level(G),calculated free energy diagram of hydrogen evolution for NbS2 and PdxNbS2 with different atomic configurations(H)[58]
原子工程对TMDs性能的调控是运用比较广泛的方法,研究表明,MoS2的催化活性主要位于其边缘,S空位、结构缺陷以及晶界的引入可以增加活性位点数量,从而用于提高催化活性,金属掺杂和插层可以活化与其相邻原子的活性.其中Zhou等[47]报道的铁磁Bowl-like MoS2利用外界垂直磁场来增加一倍的电流密度,Yang等[55]报道的利用插层转换的方法来制备具有优异催化性能的Co-Pd-MoS2,是比较新颖的研究方法,对性能也有突破性的提高,为原子工程研究提供了可行的研究思路和方法.
3.2 相工程
TMDs是层状无机化合物,通常存在H相和T相两种相[图3(A)].不同的相往往表现出不同的性质,1T-MoS2具有金属性,而2H-MoS2具有半导体性质[17,20,22],近年来,科学家们致力于通过调控TMDs的相,并使其保持稳定来提高对HER反应的催化性能.
2013年,Voiry等[24]采用锂离子插层的方法,将WS2剥落成单层且具有1T相结构的纳米片,研究表明金属1T位点是增强WS2纳米片催化活性的重要因素.之后,他们采用无溶剂插层的方法[61]剥离出具有很高浓度的1T-MoS2,并且指出1T-MoS2具有面内活性,剥落样品中金属1T相的浓度很高,从而大大改善了HER的电荷转移动力学,较之前具有更优越的性能[图11(A)和(B)].
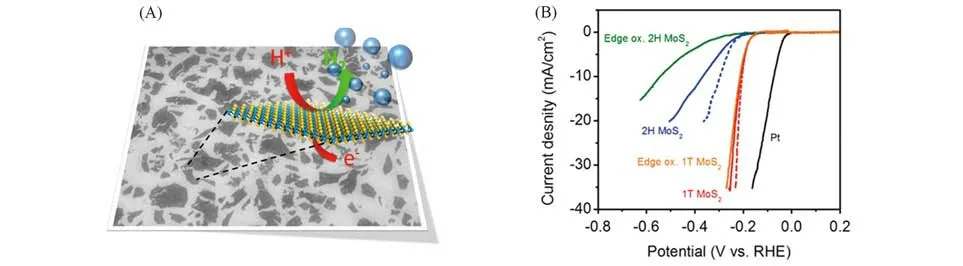
Fig.11 Electron microscope images,schematic of HER mechanism(A)and polarization curves(B)of 1T⁃MoS2[61]
除了直接合成理想相结构的TMDs,可添加额外的元素来诱导相变.如Yang等[62]通过与Re合金化获得T相MoS2,当Re>50%时,MoS2由六方相(H)转变为稳定的扭曲四方(DT)结构[图12(A)].测试结果表明,Re0.55Mo0.45S2具有低起始电位90 mV(vs.RHE)和较小的Tafel斜率(56 mV/dec),而且具有非常优越的稳定性[图12(B)],明显优于T相的MoS2[61].Qi等[57]证明单原子Co可使得MoS2从H相转变为扭曲的1T相(D-1T)[图9(H)],这是由于Co—S之间存在很强的相互作用,Co原子的锚定引起MoS2载体对D-1T的配位重建.Kwon等[36]通过水热反应合成了富硒MoSe2纳米片[图12(C)],研究表明,当Se/Mo摩尔比>2时发生了H到1T′相的转变.MoSe2.3在电催化HER方面表现出最佳性能,过电位为130 mV(vs.RHE),Tafel斜率为46 mV/dec[图12(D)].提供了更多对过量硒原子对结构和性质影响的理解,不仅诱导了H到1T′的相变,而且还提高了它的性能.
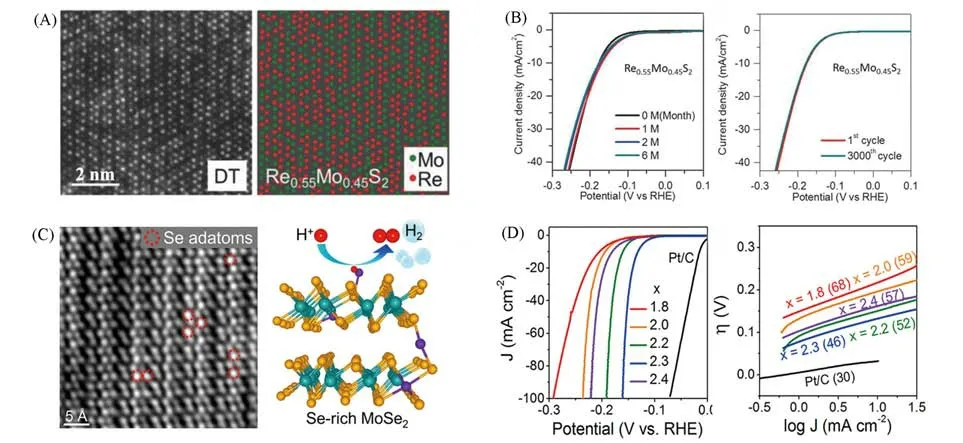
Fig.12 Structure of Re0.55Mo0.45S2 alloy monolayers(A),polarization curves and stability test of Re0.55Mo0.45S2 alloy(B)[62],HAADF⁃STEM of MoSe2.3 and DFT calculation of HER pathways(C),polarization curves and Tafel plots of MoSex(D)[36]
除了纯相TMDs的HER催化剂,多相催化剂也具有出色的HER催化表现.Wang等[63]通过水热法合成了1T/2H MoS2多相催化剂[图13(A)],研究表明1T相的存在为HER提供了更多的活性位点和更好的电导率,2H相有利于亚稳1T相的稳定化.酸性环境中,Tafel斜率为46 mV/dec,过电位也得到了优化[图13(B)和(C)].Wang等[64]报道了一种磷蒸气诱导的方法制备1T-2H MoS2的异质结构[图13(D)~(F)],面内1T-2H MoS2异质结构表现出显著改善的电导率(8630 S/m)、高度暴露的活性位点和亲水性[图13(G)],有利于电子和离子的高速传输,碱性环境中表现出良好的电催化活性:低Tafel斜率(65 mV/dec)和高的TOF(250 mV时为13.14 s—1)值[图13(H)~(J)].Zhu等[65]在实验和理论上研究了MoS2基面中的2H-1T域边界,边界处ΔGH*=—0.13 eV,非常接近Pt(111)表面[图13(K)~(M)],并且指出在酸性和碱性条件下均具有长期稳定性和通用性.
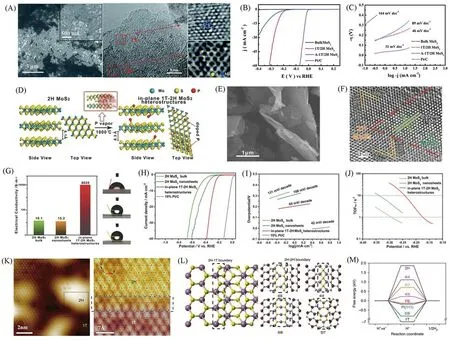
Fig.13 SEM and HRTEM images of the as⁃prepared 1T/2H MoS2(A),polarization curves(B),Tafel slopes of 1T/2H MoS2(C)[63],schematic for the partial phase transition process of semiconducting 2H MoS2 to metallic 1T MoS2 induced by phosphorus atoms(D),SEM(E)and HRTEM images(F)of in⁃plane 1T⁃2H MoS2 heterostructures,electrical conductivity and contact angle of water droplets on the surface of in⁃plane 1T⁃2H MoS2 heterostructures,2H MoS2 bulk and 2H MoS2 nanosheets,respectively(G),polarization curves(H),Tafel slopes of 1T⁃2H MoS2 heterostructures(I),comparative TOF values(J)[64],STM topography of as⁃treated MoS2 showing mixed 2H(bright)and 1T(dark)domains(K)and theoretical simulations(L,M)[65]
过渡金属硫族化合物的晶体质量和晶体结构会影响其导电性、超导性和化学稳定性等性能,通过调控晶相来调控TMDs的析氢催化性能,也是研究较广泛的调控方法.金属相具有更加优异的导电性,此外,对于第六副族TMDs,H/T相的异质结构不仅增加了材料的导电性,而且相比于纯的T相,也提高了的材料的稳定性,表现出优越的析氢催化活性.
3.3 TMDs异质结
迄今,TMDs异质结[39,40,66,67]用于电催化的研究较少,2014年,Tsai等[68]通过理论计算研究了MoS2和不同载体之间的相互作用,为研究TMDs异质结提供了重要思路.Vikraman等[69]基于之前的物理化学方法,优化合成了MoS2/WSe2异质结[70],酸性环境中,具有116 mV的过电位和76 mV/dec的Tafel斜率,并且具有20 h的稳定性[图14(A)].研究表明,制备的异质结构产生了界面电子转移,降低了电极表面对H原子的吸附能力,从而显著提高了析氢HER动力学.Chen等[71]采用界面工程方法成功构筑了富含边缘异质界面[edge1T-MoS2/edgeNi(OH)2]位点的1T-MoS2QS/Ni(OH)2催化剂(Quantum sheet,QS)[图14(B)],具有优异的碱性析氢活性:过电位为57 mV,Tafel斜率为30 mV/dec,在500 mA/cm2的电流密度下,可持续工作100 h以上[图14(C)和(D)].实验和理论研究表明,复合边缘催化位点[edge1T-MoS2/edgeNi(OH)2]具有较低的水解离能垒(ΔGH2O=0.02 eV)和氢吸附能(ΔGH*=—0.15 eV),以及适中的氢氧根吸附能(ΔGOH=—2.72 eV)[图14(B)].该研究对于高性能的碱性析氢复合电催化材料的界面结构设计具有极大的借鉴意义.

Fig.14 Structure and performance characterization of WSe2/MoS2 heterostructure on FTO substrate(A)[70],theoretical investigation of 1T⁃MoS2QS/Ni(OH)2(B),HER performance of 1T⁃MoS2QS/Ni(OH)2 in 1 mol/L KOH(C,D)[71]
异质结对TMDs析氢催化活性的研究是近几年开始研究的一种比较新颖的调控方法,两种物质的界面处具有一些特殊的物理化学性质,Chen等[71]报道的1T-MoS2QS/Ni(OH)2催化剂具有非常优异的碱性催化活性,异质结的构造为TMDs析氢催化的性能调控提供了一个新的研究思路.
3.4 非第六副族TMDs
除了第六副族的TMDs,第五副族和第七副族的TMDs也被用于作为HER的催化剂.Fujita等[72]采用锂离子插层的方法剥离了单层1T′-ReS2.Liu等[73]用CVD法合成了H-TaS2和H-NbS2晶体薄片,可随着HER的进行优化其形貌以增强电荷转移和活性位点的可及性[图15(A)],从而可以实现以最小的催化剂负载来得到优越的催化性能,测试表明,过电位为50~60 mV(vs.RHE),Tafel斜率为30~37 mV/dec[图15(B)和(C)].Shi等[74]报道了用低压CVD合成厘米级别超薄的2H-TaS2,指出其与Pt相当的催化性能来源于边缘和面内丰富的活性位点[图15(D)和(E)].Feng等[75]制备了新型的3R-TaS2,反对称性的破坏使更多的活性位点暴露出来,之后又设计出分形裂纹结构的八角状3R-TaS2[76][图15(F)],经过特殊设计的分形和裂纹树枝状结构可以暴露更多的边缘活性位点,表现出更优异的催化性能[图15(G)和(H)].Zhang等[77]使用微型电化学器件研究了各种2D TMDs的基面活性,表明3R-NbS2具有良好的HER催化性能[图15(I)~(K)],此外,Yang等[78]用CVD合成了Nb插层的Nb1.35S2,在420 mV时电流密度高达5000 mA/cm2,这一研究为2D TMD催化剂达到类贵金属的电流密度提供了重要的研究思路.这些均为非第六副族TMDs在催化中的应用开辟了途径.

Fig.15 Schematic of the proposed mechanism for the morphology change(A),polarization curves(B)and Tafel plots(C)of H⁃TaS2,H⁃NbS2[73],schematic illustration of the HER process of 2H⁃TaS2/Au foils(D),hydrogen adsorption energies at S⁃edge,Ta⁃edge,and basal⁃plane of 2H⁃TaS2,respectively(yellow,cyan,and grey balls represent S,Ta,and adsorbed H atoms,respectively)(E)[74],morpholo⁃gy of 3R⁃TaS2(left)and cracked eight⁃awn star TaS2(CE⁃TaS2)(right)(F)[75],polarization curves(G)and durability test(H)of CE⁃TaS2[76],schematic illustration of the local electrochemical measure⁃ment setup(I),the HER performance of the 2H⁃MoS2,2H⁃TaS2(J,K)[77]
4 总结与展望
以近年来的研究成果为例,总结和讨论了TMDs作为析氢催化剂的合成方法与研究进展.TMDs催化性能不佳归因于活性位点的缺乏和电导率较差.然而,一个优异的TMDs析氢催化剂应具有高密度的活性位点、优异的电荷传输能力、优异的稳定性及合成方法绿色简易且低成本等优点.基于此,可通过原子工程(硫空位、结构缺陷、掺杂合金、插层)、相工程和异质结来优化和探索新的TMDs催化剂.
原子工程主要可活化惰性基面,增加活性位点[46-58];相工程主要在于构建金属相,增强电荷转移和活性位点的数量,如第六副族TMDs的T和T′相,然而使其相结构稳定也是重要的研究内容,研究表明,晶相异质结的构建有利于相结构的稳定[63],同时晶相异质结的连接处具有优异的催化活性[64,65];原子工程和相工程往往有着很深的渊源:通过杂原子掺杂、固有元素的的富余可以诱导特定相的形成[36,62];异质结对于电催化的研究比较缺乏,但是对于研究相对较少的碱性HER催化剂具有深远的意义[71];而目前的研究仍主要集中在第六副族TMDs的催化产氢,系统研究其它TMDs的性能也将是一个充满活力的发展方向.
综上,TMDs作为析氢催化剂已经取得很大的进展,然而距离商业化还有很大的一段距离,并且大多还未达到贵金属的催化性能,构建满足所有优异特征的基于TMDs的催化剂仍然是一个很大的挑战.如Tafel斜率为29 mV/dec的MoS1.65[45],过电位为42 mV(vs.RHE)、Tafel斜率为32 mV/dec的SA Co-D 1T MoS2[57],它们都表现出类Pt的催化性能,然而其长循环稳定性仍可能是问题,且就原子缺陷和单原子而言,其位置的精确调控是一个大挑战.为了调整TMDs的HER催化性能,还需考虑其它方法,如构建Janus结构、异质结构、原子插层,这些方法目前研究相对较少,但是由于它们新奇的结构和活性位点暴露率增加,可能获得兼具高稳性与高活性的析氢催化剂,具有很大的前景.
虽然目前析氢催化剂的研究已取得了突破性的进展,但是在催化活性和循环稳定性方面仍有很大的发展空间.目前实验室的稳定性测试一般测试电流较少,离实际应用仍有一定的距离,进一步研究需要更加贴近实际应用的测试条件.通过结合理论和实验研究,合理设计催化剂,TMDs有望在析氢催化研究中替代贵金属,实现商业化.
