团簇Ti4P的催化析氢研究*
2021-02-26井润田方志刚吴庭慧
井润田,方志刚,吴庭慧
(辽宁科技大学化学工程学院,辽宁鞍山 114051)
随着经济的飞速发展以及重工业等耗能产业的崛起,环境问题日趋严重,而有效解决此类问题的方法便是绿色清洁能源的开发。氢气作为新型清洁能源,一直受到国内外众多科学家们的广泛关注。目前制取氢气的常用方法为电解水析氢,铂碳(Pt/C)催化剂在电催化析氢过程中,表现出优异的反应活性。然而,铂金属价格昂贵、储量有限,限制了其规模化应用[1]。非晶态合金材料因其优异的催化性能[2-3]、电化学性能[4]、磁学性能[5]而备受瞩目,其中钛基合金由于其优异的抗蚀性[6]、催化反应活性[7]以及光催化活性[8]被广泛关注,而高稳定合成钛基催化剂极具挑战,P的掺杂则成功解决了这一难题。原因在于Ti-P的相互作用可以稳定还原钛基化合物,大大提高钛基化合物的催化稳定性[9]。不仅如此,Ti-P系列催化剂还能提高氢气产生的选择性,有效提高了催化析氢性能[10]。因此,从Ti-P体系出发,并依据Ti和P摩尔比4∶1时具有最为优异的电循环性能[11],设计团簇Ti4P模型进行Ti-P体系的催化析氢性能研究,以期找到催化析氢性能最为优异的结构模型。
1 模型及研究方法
1.1 模型的构建和计算方法
依据拓扑学原理[12]设计出团簇Ti4P的平面以及空间构型,并在密度泛函理论[13]的基础上根据Gaussian09程序采用较高量子水平的B3LYP/Lanl2dz方法分别对二、四重态下的构型进行结构优化和频率计算及其验证,最终获得7种优化构型。计算时对Ti原子采用Fang等[14]的18-eECP的双ξ基组,对类金属P原子采用Dunning/Huzinaga双ξ基组,且对P加极化函数ξP.d=0.55[15]。
1.2 催化析氢机理
以团簇Ti4P(下文以M来表示)为对象进行模拟Ti-P体系,并对其进行系列化学反应分析及研究,其中进行催化水电解析氢反应时的反应机理分为以下两个步骤:①团簇模型吸附氢原子M+e-+H2O→M-Hads+OH-;②氢气的析出,分为2种途径,电化学解吸M-Hads+e-+H2O→M+H2+OH-,化学重组法2M-Hads→2M+H2。
2 结果与讨论
2.1 团簇Ti4P的各优化构型及稳定性分析
采用上述理论方法对团簇Ti4P进行程序运行及计算,最终得到如图1所示的7种优化构型,其中二重态3种(空间结构包括三角双锥和戴帽三角锥);四重态4种(空间结构包括戴帽三角锥、三角双锥和四棱锥)。为方便比较,以能量最低的戴帽三角锥构型1(4)作为能量零点,设其能量值为0 kJ/mol,并将各优化构型按相对能量由低到高的顺序排列(各相对能量数值已在其优化构型图下方标出),构型图下方小括号内2和4分别为所属重态。观察团簇Ti4P各优化构型,从能量角度来看,构型1(4)的能量最低,即为最稳定的优化构型。
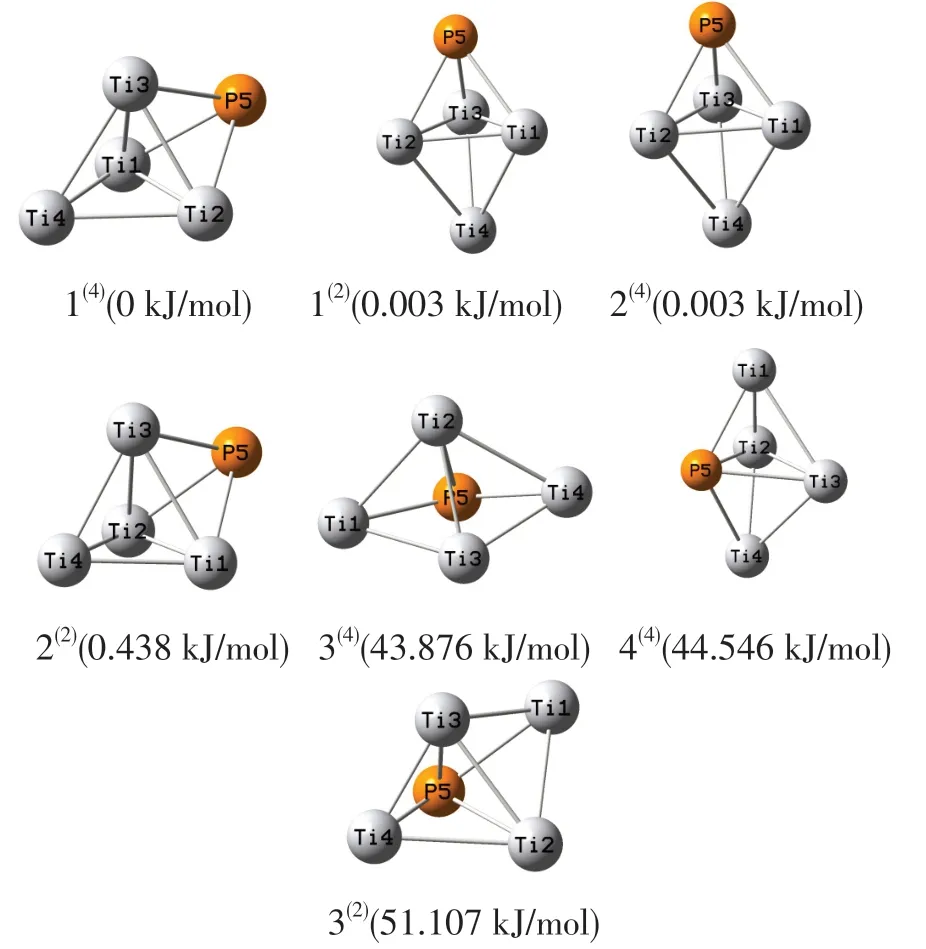
图1 团簇Ti4P的优化构型
为了更加透彻地了解团簇Ti4P各优化构型的稳定性,列出7种优化构型的校正能(EZPE)、吉布斯自由能变(ΔG)以及结合能(EBE),结果见表1。

表1 团簇Ti4P的能量参数 单位: kJ/mol
由表1可见:构型1(4)的能量最低(-626 397.289 kJ/mol),热力学稳定性最好;构型3(2)的能量最高(-626 346.111 kJ/mol),热力学稳定性最差。
为了更加直观地观察团簇Ti4P优化构型能量参数的变化趋势和参数间的关系,绘制如图2所示的能量变化趋势图。
由图2可见:团簇Ti4P各优化构型间的ΔG随着能量的升高而增大,而EBE则随着能量的升高而降低,二者恰好呈相反趋势。由于结合能表示团簇分子间结合的紧密程度,因此用结合能分析构型间的稳定性,即EBE的值越大,表明构型的相互作用较强,结合就越稳定,稳定性更好;反之,EBE的值越小,则表明稳定性越差。除此以外,判断一个反应自发进行的程度可以根据吉布斯自由能变的数值大小得到,小于零时可以自发进行反应,大于零时则恰好相反,且数值越小就越容易自发反应(小于零时)。根据图2还可以发现,团簇Ti4P的7种优化构型均能稳定存在并自发进行反应。同时随着能量的逐渐增大,各优化构型的稳定性逐渐减弱,并且自发进行的难度越来越大。

图2 团簇Ti4P各优化构型的ΔG和EBE的变化趋势
2.2 团簇Ti4P的催化析氢性能研究
2.2.1 HOMO图与水分子的LUMO图
前线电子是团簇分子的价电子,且前线电子是各化学反应过程中最早发生关键作用的电子,其所在的轨道便被称作前线轨道。其中HOMO轨道被电子占据且能量最高,LUMO轨道能量最低且没有电子在该轨道。根据以上分析HOMO轨道以及LUMO轨道可以判断是否发生反应,同时也可作为判断团簇反应活性的重要依据。因此,对团簇的前线轨道进行研究是有必要的。团簇模型中的电子从HOMO轨道转移至水分子的LUMO轨道便是进行催化析氢反应的第一步,这一步是各构型与氢原子间的吸附,即形成计算方法部分所提到的M-H结构模型。为了得到团簇模型吸附氢原子时电子转移的规律,绘制出图3。为了能够更加全面具体的分析该规律,因此根据其存在的2种电子(自旋向上的α电子和自旋向下的β电子)分别展开讨论。

图3 团簇Ti4P各优化构型的HOMO图和H2O的LUMO图
由图3可见:团簇Ti4P 7种优化构型的HOMO图和水分子的LUMO图均存在着深浅2种颜色的区域,二者分别代表正负2种不同相位的轨道波函数,深色为负相位,浅色为正相位,且两者的形成均是因为电子出现的概率较大,即电子云密度较大,同时说明这2种区域在进行化学反应时较为活跃。
根据前线轨道理论[16],只有当HOMO、LUMO两轨道同号重叠时才能有效完成电子的转移以及成键,因此在进行吸氢反应时,团簇Ti4P各优化构型的HOMO轨道的波函数相位只有与水分子的LUMO轨道波函数相位相同时反应才能发生。根据水分子的LUMO图可知,水分子LUMO轨道几乎完全为深色区域,因此水分子外围轨道为负相位。根据前文分析可知:只有在团簇Ti4P 7种优化构型的HOMO轨道外围也为负相位时二者发生化学反应的概率才最大。观察各优化构型的HOMO图可以发现,相较于β-HOMO图,α-HOMO图的深色区域要更大且均分布在外围,因此α-HOMO与水分子的LUMO轨道同号重叠的概率更大,即更容易发生化学反应,说明团簇Ti4P在进行吸氢反应时,α-HOMO比β-HOMO更具优势,贡献更大。
2.2.2 团簇Ti4P与水分子之间的轨道能级差
为了能够更加清楚地分析判断出团簇Ti4P和水分子在进行吸附氢原子时的难易程度,列出团簇Ti4P 7种优化构型的HOMO轨道与水分子LUMO轨道之间的能级差,见表2。
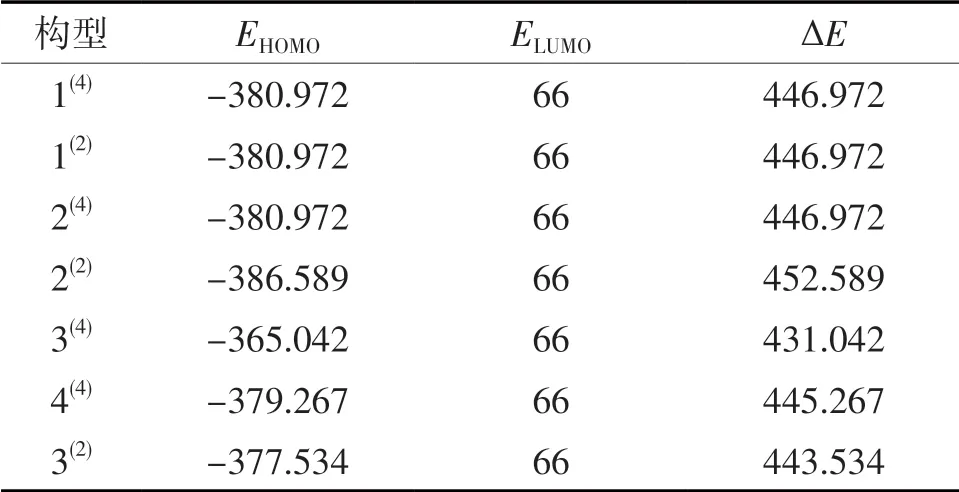
表2 团簇Ti4P的HOMO轨道能级与水分子的LUMO轨道能级之差 单位:kJ/mol
由于2种物质的HOMO与LUMO轨道间的能级差小于579 kJ/mol时才能发生电子间转移[18],即发生化学反应。据此可知,当团簇Ti4P的HOMO轨道能级与水分子的LUMO轨道能级之差小于579 kJ/mol时,电子才能从优化构型的HOMO轨道转移至水分子的LUMO轨道。根据表2数据可知,团簇Ti4P的7种优化构型的ΔE均小于579 kJ/mol,由此推断在进行吸氢反应时7种优化构型均具有良好的反应活性。并且构型3(4)的ΔE值最小(431.042 kJ/mol),因此该构型电子最容易从其HOMO轨道转移至水分子的LUMO轨道,即最容易发生吸氢反应。综合所有数据来看,即使是ΔE数值最大的构型2(2),其与构型3(4)也仅相差21.547 kJ/mol,再结合图3来看,7种优化构型HOMO图中的深色区域均相对较大,且分布在外围。综上所述,团簇Ti4P的7种优化构型在进行吸氢反应时反应活性均较为良好。
2.3 (Ti4P)-H的解吸过程
为更加清楚地了解团簇Ti4P的催化析氢反应过程,对催化析氢的步骤二即析出氢气的过程进行系统分析,团簇Ti4P各优化构型吸附氢原子后的模型表示为(Ti4P)-H型,如图4所示,7种优化构型的氢原子均吸附在金属原子上,说明团簇Ti4P各优化构型中的Ti原子即为催化反应的潜在活性位点。为更加清楚地分析出团簇Ti4P在吸附氢原子后的稳定性,将(Ti4P)-H 7种优化构型的校正能列于表3,其中E0表示加氢前后校正能间的差值。
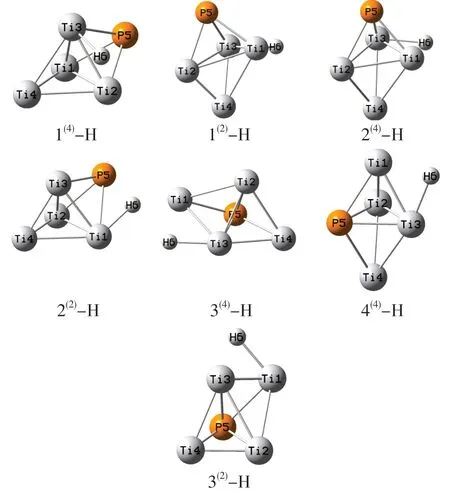
图4 (Ti4P)-H的优化构型

表3 (Ti4P)-H 7种优化构型的校正能 单位: kJ/mol
将表3与表1数据进行对比可以发现,加氢以后的团簇模型校正能有了明显的下降,同时说明加氢后的构型整体更加稳定。加氢后校正能数值最小的为构型2(4)(-628 000.569 kJ/mol),而未加氢时校正能最小的为构型1(4)(-626 397.289 kJ/mol)。结合图1和图4,可以发现构型2(4)在加氢后构型变化较为明显,而构型1(4)的空间构型以及原子位置均无明显变化且二者氢原子均加在了Ti3原子上,这在一定程度上也说明原子位置的变化是影响构型稳定性的重要因素。另外,对于加氢前校正能最大的构型3(2)来说,加氢以后校正能要低于构型2(2)且差值较大(E0=1 548.620 kJ/mol),结合图1和图4可知,加氢后构型3(2)空间构型变化较大,金属Ti原子位置变化较为明显,而构型2(2)变化较为明显的为P原子,推断金属Ti原子的位置对构型稳定性的影响要大于非金属P原子。另外通过观察E0数值可以发现,四重态的4种构型E0值要明显大于3种三重态构型,结合图4可以发现,4种四重态构型氢原子均附于Ti3原子上,而其余3种优化构型则是氢原子附于Ti1原子上,说明Ti1、Ti3原子更容易吸附氢原子,是潜在的催化活性位点,同时也说明氢原子加在Ti3原子上会使得团簇整体的稳定性提高。
2.3.1 (Ti4P)-H 的电化学法析出氢气
在进行化学吸附后,吸附氢原子的(Ti4P)-H 团簇模型将与水分子的另一氢原子继续结合,且两氢原子将以氢气的形式脱离团簇模型,析出氢气的模型则回归Ti4P结构,团簇的催化作用发挥完毕。由于(Ti4P)-H 析出氢气的过程与团簇Ti4P吸附氢原子的过程相类似,区别在于反应底物由Ti4P变为(Ti4P)-H,因此可以根据(Ti4P)-H 各构型的HOMO能级与水分子的LUMO能级间的能级差来判断(Ti4P)-H各构型析出氢气的难易程度,具体数值如表4所示。

表4 (Ti4P)-H 7种优化构型的HOMO能量与H2O的LUMO能级间的能级差 单位:kJ/mol
由表4可见:整体上看,团簇Ti4P的7种优化构型在加氢以后HOMO轨道与水分子的LUMO轨道间的能级差仍小于579 kJ/mol,说明(Ti4P)-H 各构型在进行电化学析出氢气时仍然具有良好的催化活性。根据表4数据可以看出,除构型2(2)和4(4)外,其余5种构型在加氢以后能级差均有所增加,说明这5种构型在加氢后与水分子反应时的反应活性减弱,而构型2(2)和4(4)则与之相反。构型2(2)加氢以后HOMO能级与水分子的LUMO能级之差最小,进行电化学析出氢气时的反应活性也最优异。
2.3.2 (Ti4P)-H的化学重组法析出氢气
依据化学重组法分析(Ti4P)-H析出氢气,分子中的2个氢原子作为中间体相互结合后成为氢气。(Ti4P)-H模型析出氢气后则重新转变为初始的Ti4P结构。据此分析,只有当团簇Ti4P模型分子中的氢原子结合强度较小时氢气才更容易析出。为了更加清楚地分析化学重组法析出氢气的情况,将(Ti4P)-H 7种优化构型的结合能数据列于表5。

表5 (Ti4P)-H 7种优化构型的结合能 单位:kJ/mol
由表5并且结合表1数据可见:此时的结合能数值要远高于初始优化构型的结合能数值,这说明吸附氢原子的结构模型若通过化学重组法再继续吸附氢原子是较困难的。由于构型2(2)的结合能增值以及该构型(Ti4P)-H模型结合能数值均为最小,说明该构型最容易通过化学重组法继续结合氢原子,同时也说明该构型通过化学重组法的催化反应活性较好。
3 结论
依据拓扑学原理和密度泛函理论,设计出可能存在的构型并且对其进行量子化学计算。得到优化构型后对其稳定性进行分析,根据催化析氢反应机理,找到最优异的构型,据此得到以下结论。
1)分析团簇Ti4P的7种优化构型的稳定性可知:构型1(4)的热力学稳定性最好,构型3(2)的热力学稳定性最差。
2)分析团簇Ti4P的催化析氢反应机理中吸附氢气和析出氢气2个过程可知:根据7种优化构型的HOMO图以及水分子的LUMO图、团簇Ti4P与水分子之间的轨道能级差,在团簇Ti4P进行吸附氢气的过程中,7种优化构型均具有较好的反应活性;根据电化学法和化学重组法可以得到(Ti4P)-H模型在析出氢气时,构型2(2)具有最好的反应活性。此外,由于构型2(2)在吸附氢气和析出氢气2个反应过程中均具有良好的反应活性,因此可以推断该构型在进行催化析氢反应时为最优异的结构模型。
