蛋白质谷氨酰胺酶的重组表达与发酵条件优化
2021-02-25李静竹胡梦君张建华
李静竹,胡梦君,张建华
(上海交通大学 农业与生物学院,上海,200240)
植物蛋白相较动物蛋白有较多的营养素和更少的卡路里,因此近年来以植物蛋白为原料的相关食品深受健康人士的推崇[1]。然而核桃、杏仁等植物蛋白饮品在加工过程中易受pH和化学添加剂等因素的影响,从而出现不同程度的分层、絮凝、沉淀等现象[2]。尽管在偏酸性液态食品中pH与植物蛋白的pI接近是诱因之一[3],但其主要原因是大多数植物蛋白含有大量的谷氨酰胺残基和天冬酰胺残基,易形成氢键使得蛋白质聚集沉淀[4],进而影响蛋白的乳化性、气泡性和凝胶性等功能性质,降低产品品质[5]。为有效解决植物蛋白生产加工中的上述难题,一般采用脱酰胺的方法对植物蛋白进行处理。
目前,工业上广泛采用酸碱湿热法和酶法对植物蛋白进行脱酰胺处理。酸法处理常会涉及高温,易引起蛋白质的水解,从而产生不良风味,影响产品品质。酶法处理可以有效脱去大豆蛋白和燕麦蛋白等植物蛋白的谷氨酰胺残基上的氨基,提高蛋白的溶解度。常用的酶有胰凝乳蛋白酶[6]、转谷氨酰胺酶[7]和肽谷氨酰胺酶等,但这些酶有一定的局限性,如转谷氨酰胺转氨酶底物专一性不强[8],肽谷氨酰胺酶对大分子蛋白质作用甚微[9]。因此,专一性强和底物特异性好的蛋白质谷氨酰氨酶(protein-glutaminase,PG)引起了越来越多的关注。
PG最初是在2001年被日本学者YAMIGHCHI等[10]从土壤中的解脘金黄杆菌发酵液分离得到。PG一般以含有信号肽的前体形式表达,需经酶切为分子量约20 kDa的成熟酶分子后才有活性[11]。研究发现PG对羟基苯氧基-谷氨酰胺-甘氨酸(Cbz-Gln-Gly)和酪蛋白等底物有脱酰胺活性[10],且不会造成蛋白质的水解,底物专一性很强,安全有效。PG对米谷蛋白[11-12]、燕麦蛋白[13]等均有良好的脱酰胺作用,且处理后其起泡性、凝胶性和溶解度等功能性质均得到一定改善。目前PG已作为国家批准的食品添加剂,但其生产菌解脘金黄杆菌发酵液中PG的酶活力仅为0.258 U/mL[10],难以工业应用,因此提高菌株产PG的能力非常重要。
对PG重组表达已有诸多研究,KIKUCHI等[14]置换PG信号肽,构建谷氨酸棒杆菌(Corynebacteriumglutamicum) pPK4:pro-mPG表达体系,表达的带前导肽(leader peptide,pro)的PG经丝氨酸蛋白酶酶切后成功获得成熟肽(mature peptide,mPG);汪正华等[15]通过重叠延伸PCR法合成PG的全长基因,构建BL21/pET32a(+):mPG表达体系,经诱导后得到以包涵体的形式表达的mPG,再经变性、复性处理后测得该mPG的酶活力为0.371 U/mL。以上研究构建的菌株或需IPTG诱导表达,或产酶量不高,无法达到商业化生产的要求。因此,构建可以较高效表达此酶且无需诱导剂的表达体系尤为重要。
本文以大肠杆菌-枯草芽孢杆菌穿梭质粒pHT43为表达载体,并将其诱导型启动子Pgrac替换为强组成型启动子P43[17-18],构建重组质粒pHT43:pro-mPG(P43),将其转化至BL21(DE3)[19]中,实现pro-mPG的胞内可溶性表达。采用胰蛋白酶对pro-mPG酶切,获得具有一定酶活力的成熟mPG。通过培养基配方和培养条件优化,进一步提高酶活。
1 材料与方法
1.1 材料与试剂
大肠杆菌Top10,BL21(DE3)为本实验室保藏;质粒pHT43,淼灵生物有限公司;pHT43:pro-mPG(P43)为本实验构建。
胰蛋白胨、酵母提取物、琼脂糖、氨苄西林,日本OXIDE公司;UNIQ-10质粒提取试剂盒、胶回收试剂盒、丙烯酰胺、3-Truecolor预染蛋白Marker、限制性内切酶KpnI和SmaI、T4连接酶、脱脂奶粉、10×TBST、异丙基-β-D巯基半乳糖苷,上海生物工程有限公司;MixPCR、核酸染料、200 bp Marker 购自TaKara;His-tag单克隆抗体、羊抗鼠抗体、ECL显色发光试剂盒,上海翊圣生物有限公司;其他培养基及有机、无机试剂均为国产分析纯。
1.2 仪器与设备
电子天平,瑞士Mettler Toledo公司;超纯水系统,美国Mili-Q公司;高压灭菌锅,日本Tomy公司;恒温培养摇床,深圳市显控自动化技术有限公司;微量台式离心机,德国Eppendorf公司;超声波细胞破碎仪,SONICS公司;超微量分光光度NanoDrop 2000c,美国Thermo Forma公司;PCR仪, Bio-Rad Laboratory;微波炉,韩国LG公司;酶标仪,TECAN;电泳仪,Bio-Rad Laboratory;转膜仪,Bio-Rad Laboratory;显影仪,ChemiDoc XRS+。
1.3 方法
1.3.1 PG基因的片段合成设计及其表达载体的构建
利用pHT43构建表达质粒。选用强组成型启动子P43替换pHT43上的原有的启动子Pgrac[20], P43序列后紧接着pHT43中自带的核糖体结合位点(ribosomebinding site,RBS)和信号肽SamyQ序列,其后为编码PG的前导肽和成熟肽序列(Genbank:AB046594),并在成熟肽N端添加6个组氨酸,片段全长1 359 bp,由上海派森诺基因科技公司人工合成。选择质粒上的KpnI和SmaI作为酶切位点,将合成的目的片段与被KpnI和SmaI酶切的pHT43通过T4连接酶连接,构建重组质粒pHT43:pro-mPG(P43)。双酶切及酶连接体系参照韦雪芳等[21]的方法。
1.3.2 PG基因的克隆及其质粒酶切鉴定
将重组质粒pHT43:pro-mPG(P43)及pHT43空载分别转化至E.coliTop10感受态细胞中,加入600 μL的LB液体培养基,在37 ℃、200 r/min培养1 h。取出复苏的Top10感受态细胞并接种于含100 μg/mL氨苄西林的5 mL LB液体培养基中,37 ℃过夜培养。次日提取质粒并进行酶切验证。
1.3.3 PG基因重组表达菌E.coliBL21的转化及其阳性转化子的鉴定
将重组质粒和空载分别转化至E.coliBL21(DE3)感受态细胞中,转化方法与培养条件同1.3.2。将复苏液均匀涂布于含100 μg/mL氨苄西林的LB平板上,37 ℃过夜培养。次日挑取平板上的单菌落转接至1 mL LB(含100 μg/mL氨苄西林)液体培养基中,37 ℃、200 r/min培养12 h。取500 μL加热煮沸15 min,作为DNA模板进行PCR扩增检测。利用生物软件oligo设计上下游引物,分别为F2499:5’-CTTTCAGCCGACT CAAACATCA-3’;R3013:5’-GTAAGGTATAAACTTTTCAGTTGCAGA-3’。
PCR反应体系为:菌液DNA模板1 μL,上下游引物各1 μL,Mix master 12.5 μL,加无菌水补足至25 μL。扩增反应条件:94 ℃预变性2 min,95 ℃变性15 s,60 ℃退火30 s,72 ℃延伸30 s,共30个循环,72 ℃延伸5 min。将目的条带切胶回收并测序。测序完全正确的阳转子-80 ℃保存备用。
1.3.4E.coliBL21阳转子的诱导表达与发酵液超声处理
将重组菌和空载菌株在LB平板上划线活化。挑取单菌落,接种于4 mL含100 μg/mL氨苄西林的LB液体培养基,在37 ℃、200 r/min过夜培养。以5%接种量将上述两种菌液分别转接至LB液体培养基中(100 μg/mL氨苄西林),于37 ℃、200 r/min摇床培养至OD600为0.8左右开始计时,分别取上述菌株培养4、8、24 h的菌液各1 mL,4 ℃、10 000 r/min离心2 min,弃去上清,用1×PBS(pH 7.2)缓冲液洗涤菌体沉淀2次。再加入500 μL的1×PBS充分重悬菌体,将其置于冰上进行超声破碎(功率为40%,10 s/10次)。超声后菌液于4 ℃、8 000 r/min 离心1 min,沉淀用1×PBS溶解。采用Nanodrop测定重组菌的超声破碎全菌液、上清液及沉淀的蛋白浓度。
1.3.5E.coliBL21阳转子的SDS-PAGE分析、镍柱纯化以及Western blot验证
将蛋白样品浓度调整为5~6 mg/mL,与4×SDS蛋白上样缓冲液按体积比3∶1混合、煮沸,进行SDS-PAGE凝胶电泳:设置恒压80 V,待样品中的溴酚蓝到达浓缩胶与分离胶的界线时,将电压调至120 V。采用NTA-Ni柱对蛋白样品进行纯化,采用不同浓度的咪唑溶液(20、50、100、250 mmol/L)分步洗脱,间接取样进行SDS-PAGE分析,收集含有单一目的蛋白的洗脱液进行脱盐,用1×PBS(pH 7.2)缓冲液替换洗脱液中的咪唑,浓缩后通过BCA法测定其蛋白浓度。参照陈俊等[22]的方法对未经考马斯亮蓝染色的PAGE胶进行湿法转膜,恒流116 mA条件下电转40 min。Western-blot相关操作参照HU等[23]的方法,采用ECL显色发光试剂盒对膜进行孵育,显影仪自动曝光1 min,观察免疫印迹结果。
1.3.6 酶活测定条件确定以及酶学性质分析
含前导肽的PG先用胰蛋白酶消化,消化试验采用何灿等[24]的方法进行适当修改,具体如下:配制不同质量浓度的胰蛋白酶稀释液(0.03、0.1、0.3、0.9、2.7 mg/mL),与纯化后的pro-mPG按1∶10混匀,消化不同时间(0~50 min,每间隔10 min),进行SDS-PAGE分析和酶活力测定。根据SDS-PAGE条带灰度分析和酶活力测定结果确定胰蛋白酶的最佳消化时间。
酶活力测定方法在汪正华等[15]的基础上进行部分调整,将消化后的酶液与底物反应不同时间(0~30 min,每间隔5 min),进行靛酚显色,确定消化酶液与底物的最佳反应时间。根据酶活力结果,选择以100 μL 酶液,用0.03 mg/mL胰蛋白消化10 min,底物反应时间20 min为酶活测定条件。
对纯化的PG进行酶学性质分析,分别在不同pH(3.5~9.5)和不同温度(25~70 ℃)下,将mPG孵育60 min后测定酶活力,探究PG的最适酶反应条件。
1.3.7 优化培养基配方和发酵产酶条件
以添加质量分数0.4%的谷氨酰胺[25]的LB培养基为基础,选用不同碳源(葡萄糖、蔗糖、麦芽糖、淀粉、山梨糖醇、甘油)替代LB中80%酵母提取物,不同氮源(牛肉浸膏、蛋白胨、酵母浸粉、大豆蛋白胨、安琪酵母粉、甘氨酸)替代LB中80%胰蛋白胨,分别添加质量分数为0.1%和0.3%的金属离子(K+、Mg2+、Ni2+、Mn2+、Cu2+、Fe2+、Ca2+)[26],探究不同碳、氮源和金属离子对产酶的影响,并选择不同的温度(25 ℃、30 ℃、33 ℃、37 ℃、40 ℃)和不同的初始pH(5.0、5.5、6.0、6.5、7.0)分别培养重组菌,探究培养条件对产酶的影响。
1.3.8 数据分析
酶的纯化及发酵条件优化等实验均进行3次重复,采用prism统计软件进行数据处理及显著性分析。
2 结果与分析
2.1 目的片段的合成与重组载体的构建
由于产PG的解脘金黄杆菌与大肠BL21亲缘关系较远,为实现PG在BL21中的有效表达,本研究不使用编码PG自身的信号肽[20,27],而是利用大肠杆菌-枯草芽孢杆菌穿梭质粒pHT43及其本身含有的信号肽SamyQ构建重组质粒。图1,为本研究构建的重组质粒pHT43:pro-mPG(P43)。
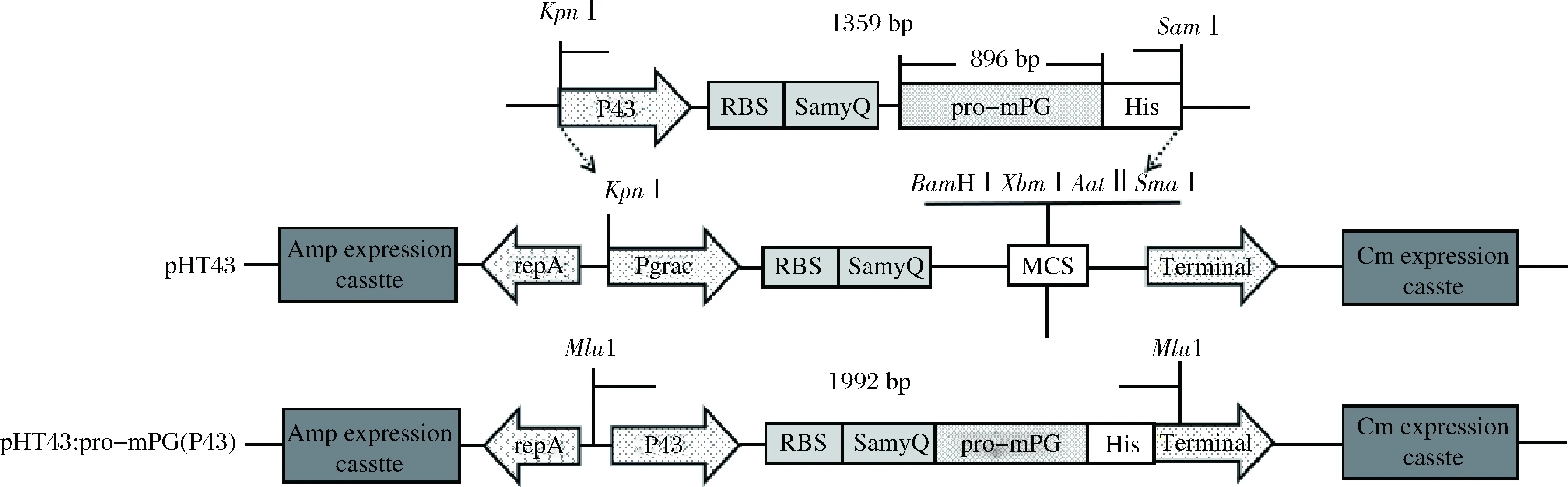
图1 pHT43:pro-mPG(P43)重组质粒图Fig.1 Schematic diagram of recombinant plasmidpHT43:pro-mPG(P43)
1.2 PG基因的克隆菌和重组表达菌的转化及鉴定
将重组质粒转化至Top10感受态细胞中,对质粒进行双酶切验证。结果如图2-a所示,泳道1为重组质粒经MluI酶切后得到的片段,其片段大小与预期的7 288 bp和1 827 bp相符。将疑似阳性的重组质粒进行测序,与目的序列一致性为100%,证明成功构建了PG基因的克隆菌株。同理,提取Top10克隆菌株的重组质粒转化至BL21(DE3)感受态细胞中,采用F2499/R3013引物对BL21转化子进行菌液PCR验证。结果如图2-b所示,阳性转化子PCR产物与阳性对照(重组质粒)条带大小一致,在1 300 bp附近有单一的特异性扩增条带,将PCR产物胶回收后并进行测序验证,经BLAST比对结果显示一致性为100%,证实PG重组表达菌株构建成功。
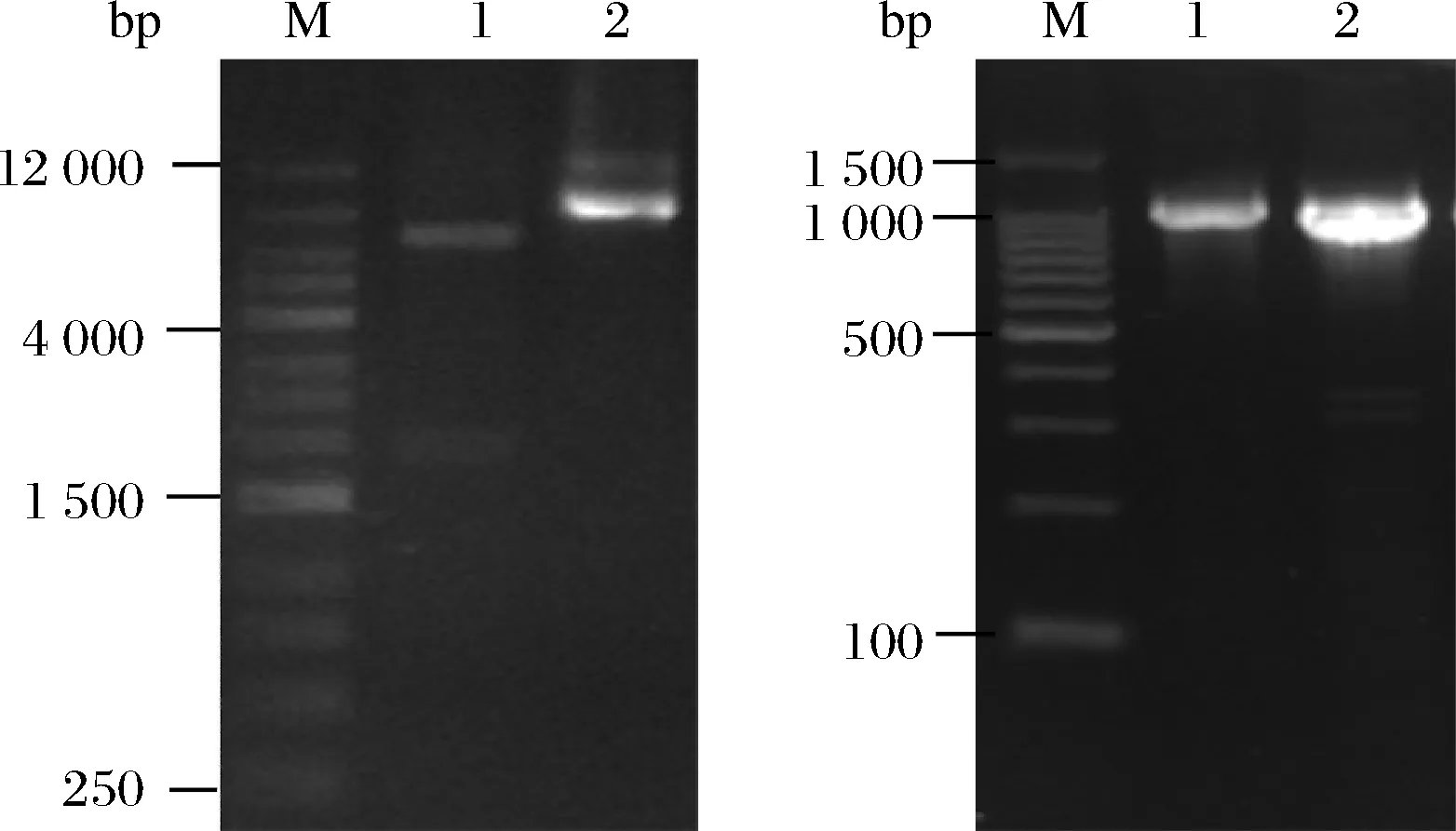
a-pHT43:pro-mPG(P43)质粒酶切b-阳性菌液PCR扩增产物a:M-1kbMarker,1-酶切后质粒,2-未切割质粒;b:M-100 bp Marker,1-阳性转化子PCR产物,2-阳性对照图2 pHT43:pro-mPG(P43)克隆及PCR验证Fig.2 Verification ofpHT43:pro-mPG (P43) clone and recombinant transformant by PCR
1.3 pro-mPG的表达、纯化及Western blot验证
对菌体破碎上清液进行SDS-PAGE分析,结果如图3-a所示,35 kDa上方出现一条带,与DNAMAN软件预测的pro-mPG分子量37.54 kDa相符,可能为成功表达的可溶性pro-mPG。镍柱纯化的结果如图3-b所示,结果表明,蛋白可以被镍柱吸附,在20 mM的咪唑下可被洗脱,且能够与鼠抗组氨酸标签抗体特异性识别并产生免疫印迹,因此确认该蛋白为pro-mPG。

a-pro-mPG的表达及纯化b-Western blot验证图a:1、2-重组菌全菌液,3-重组菌1的超声破碎上清液,4~7-20 mmol/L咪唑洗脱液,8、9-250 mmol/L咪唑洗脱液;b:1、3-20 mmol/L咪唑洗脱液,2、4-250 mmol/L咪唑洗脱液,M-蛋白Marker图3 菌体破碎液及镍柱洗脱液中pro-mPG的SDS-PAGE图及Western blot验证Fig.3 SDS-PAGE image and Western blot verification of pro-mPG in the disrupted cells and elute from nickel column
1.4 pro-mPG的消化及其产物分析
pro-mPG经胰蛋白酶消化的结果如图4-a 显示,消化后出现两条低分子量(约20、30 kDa)的条带,与刘英杰报道的一致[28]。在消化时间0~50 min内,随着反应的进行,pro-mPG逐渐由37.54 kDa降解到20 kDa,且20 kDa处的条带在消化10 min后灰度最深。该蛋白疑似为切割掉前导肽后的成熟PG酶(mPG)。酶活力测定结果表明消化10 min的PG酶活力最高为0.250 U/mL。消化10 min后的酶液经镍柱纯化,得到单一条带(图4-b),Western-blot分析发现该条带同样能与鼠抗His-tag产生特异性的免疫印迹反应,确认为mPG。
1.5 mPG的酶学性质分析
图5-a为不同pH条件下测定纯化mPG酶活力的结果,发现pH 6.5时,酶活最高。不同温度下的PG酶活力结果如图5-b所示,在45 ℃条件下出现最高酶活力。且随着温度的升高,酶活力呈现先上升后下降的趋势,在70 ℃时几乎丧失活性。
2.6 PG发酵条件优化
2.6.1 不同碳源、氮源对工程菌产酶的影响
对照组采用粗酶液进行测定,酶活力为0.417 U/mL,比纯化的PG测得的酶活力高,这可能由于背景值较高以及谷氨酰胺添加的影响所致。由6-a可知,以蔗糖和麦芽糖为主要碳源的培养基产酶高于对照组,其中蔗糖组差异显著(P<0.05),最高酶活为0.490 U/mL,这可能是由于菌株对不同碳源的利用度不同导致产pro-mPG的量有差异。而以麦芽糖和淀粉为主要碳源的培养基对pro-mPG酶活力的影响不大,山梨糖醇和甘油对PG的酶活力的抑制作用显著(P<0.05)。
a-胰蛋白酶消化pro-mPGb-消化产物Western blot分析a:M-蛋白Marker;1~6-胰蛋白酶消化不同时间(0、10、20、30、40、50 min)的pro-mPG;b:M-蛋白Marker 1、2-镍柱纯化后的mPG的SDS-PAGE图,3、4-Western blot分析图4 胰蛋白酶水解的pro-mPG的SDS-PAGE图及Western blot分析Fig.4 SDS-PAGE image and Western blot verification analysis of trypsin hydrolysis of pro-mPG
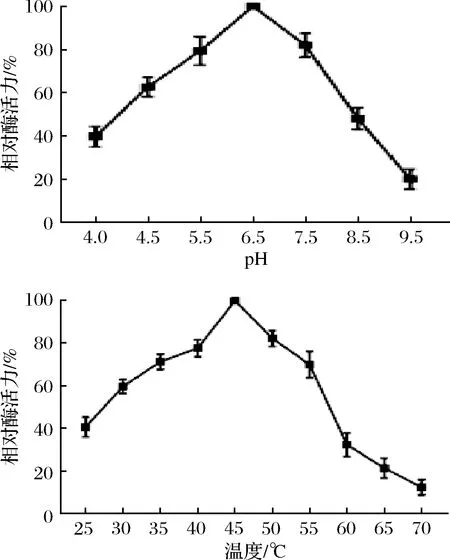
图5 pH和温度对PG酶活力的影响Fig.5 Effect of pH and temperature on PG activity
在以蔗糖为主要碳源的培养基中,考察不同氮源对产酶的影响,结果如图6-b所示,酵母浸粉对重组菌产酶影响显著(P<0.05),酶活在碳源优化的基础上进一步提高至0.603 U/mL。
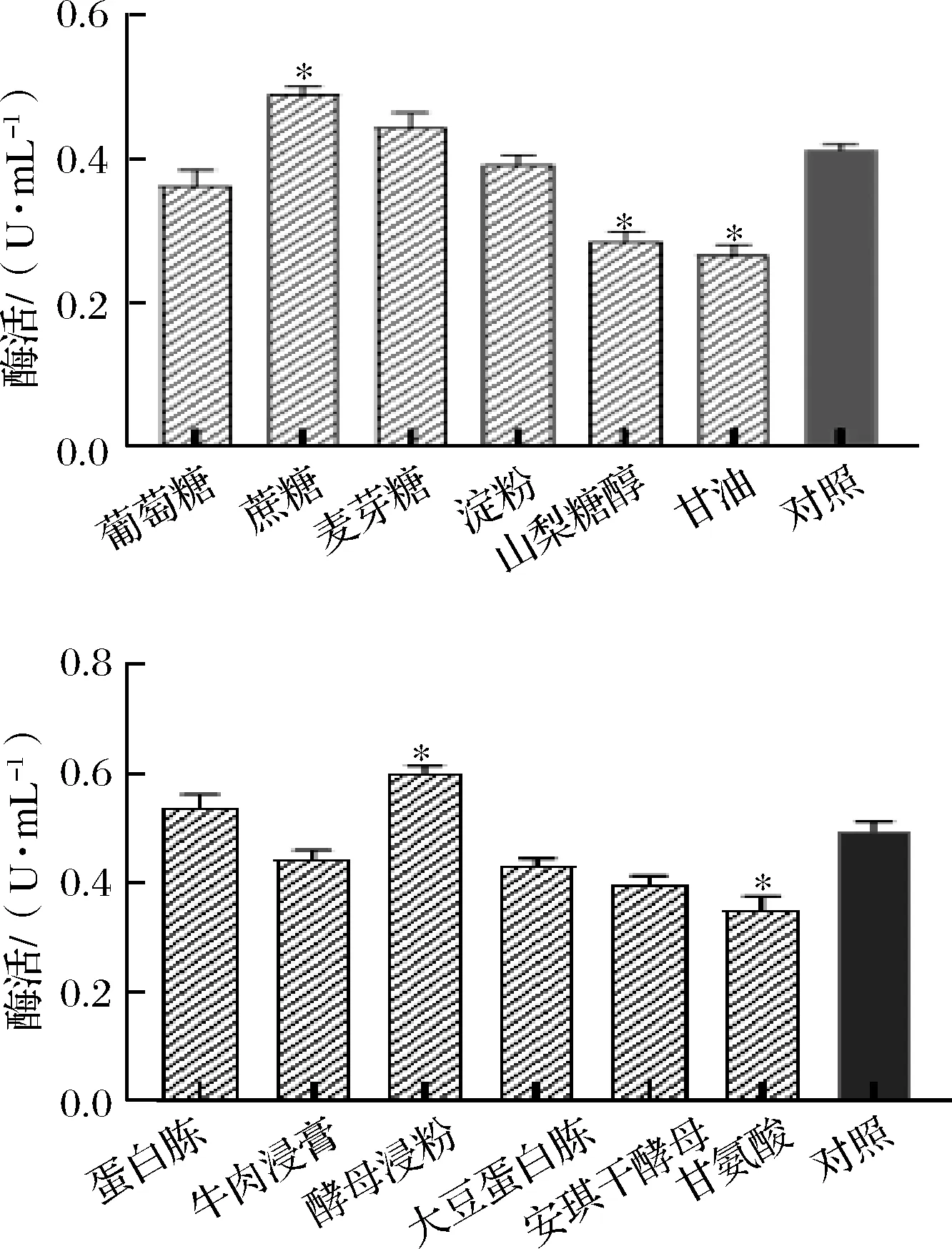
a-碳源; b-氮源图6 不同碳源和氮源对工程菌产酶的影响Fig.6 Effect of different carbon and nitrogen sources on the pro-mPG production from engineering bacteria
2.6.2 金属离子对工程菌产酶的影响
在上述最佳碳源和氮源的基础上,考察不同浓度的金属离子对产酶的影响。结果如图7所示,添加Mn2+和Ca2+均对产酶有促进作用(P<0.05),而K+、Ni2+和Mg2+对产酶的影响不显著(P>0.05),Cu2+和Fe2+的添加反而抑制了产酶。添加质量分数0.3%的Ca2+培养基最高酶活力为0.760 U/mL,与对照组相比,提高了0.157 U/mL左右。
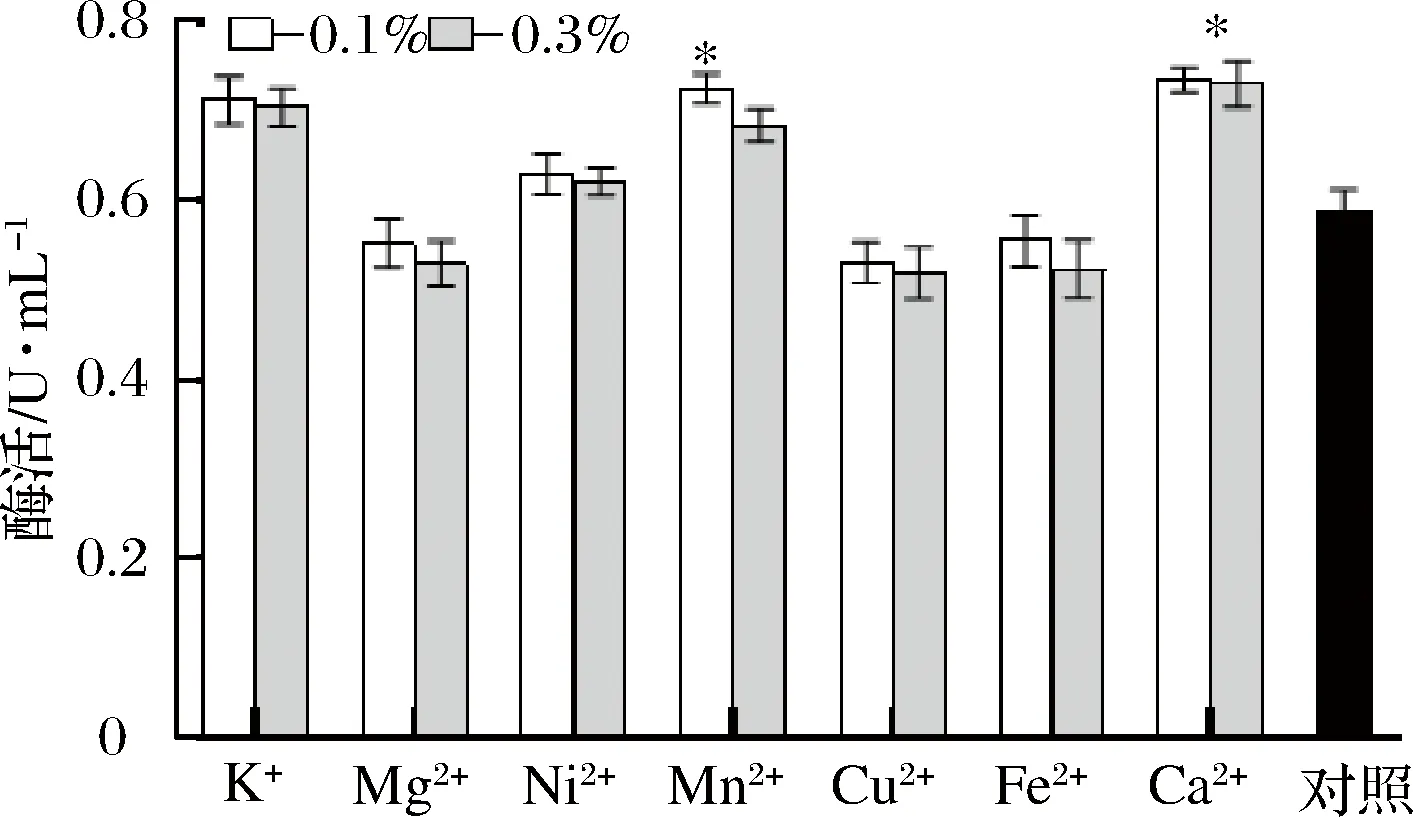
图7 不同金属添加量对工程菌产酶的影响Fig.7 Effect of different metal additions on the production of pro-mPG from engineering bacteria
2.6.3 培养条件对pro-mPG酶活力的影响
25 ℃、30 ℃、33 ℃、37 ℃、40 ℃条件下工程菌产PG的酶活力分别为0.333 U/mL、0.481 U/mL、0.623 U/mL、0.764 U/mL、0.640 U/mL,37 ℃为最适温度,如图8-a所示。不同pH条件下测得的酶活力差异不显著(P>0.05),说明pH对工程菌产PG影响较小,如图8-b所示。
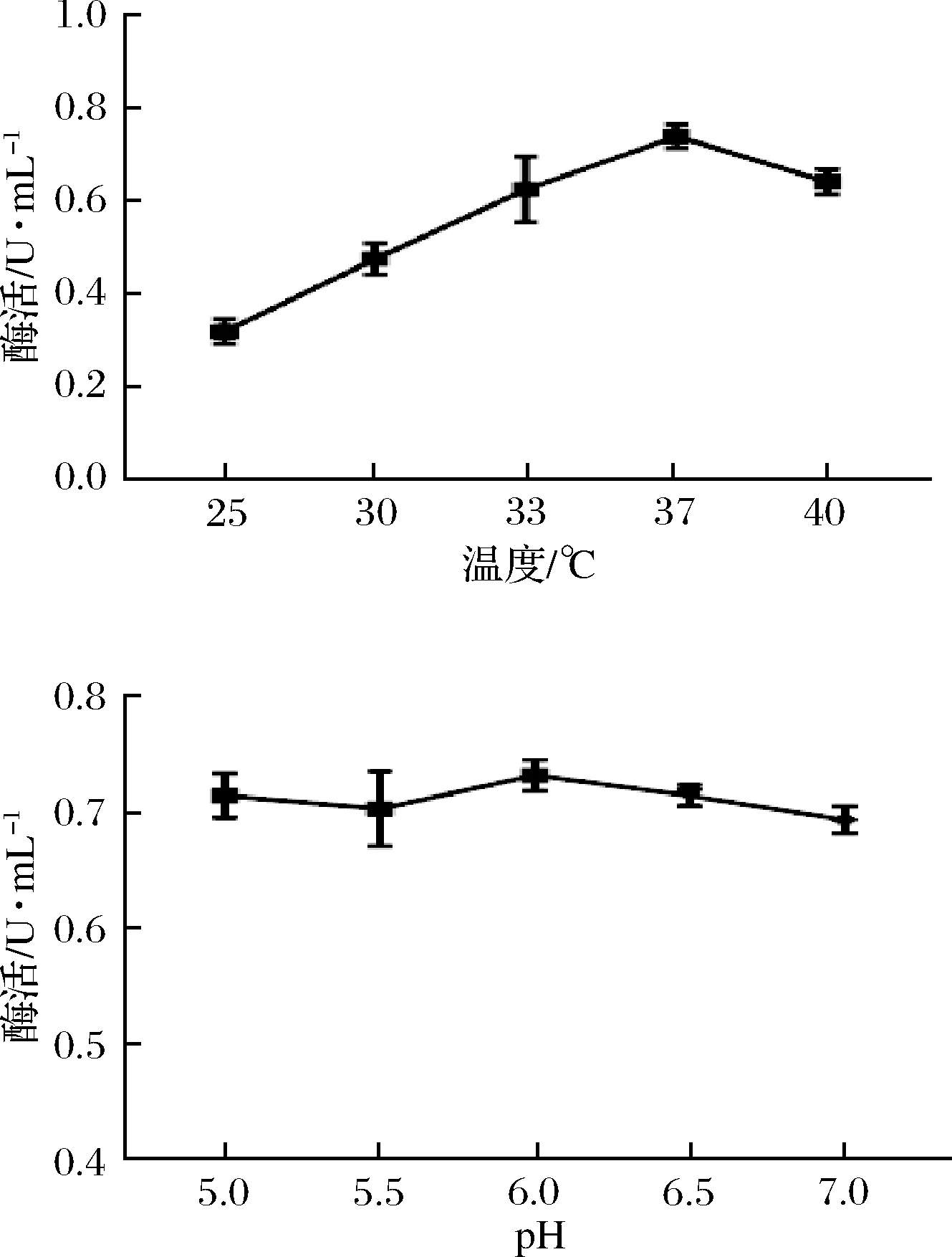
图8 温度和pH对工程菌产酶的影响Fig.8 Effect of temperature and pH on the production of pro-mPG from engineering bacteria
3 讨论
目前PG已作为国家批准的食品添加剂[29],其生产来源仍然依赖解脘金黄杆菌[16],但是该原始菌发酵产酶量少,限制了该酶的工业化生产应用。现有的工程菌过表达PG的研究,大多采用IPTG作为诱导剂[30]。但IPTG价格较昂贵,且对微生物和人体有一定毒性,故减少或避免其使用对食品酶的表达有重要的现实意义。因此,本研究采用强组成型启动子P43代替pHT43本身的诱导型启动子Pgrac,利用细菌的群体感应表达PG,不使用IPTG,提高了目标酶在食品中应用的安全性。
由于PG在植物蛋白脱酰胺方面有良好的应用前景,近年来,重组表达PG的相关研究已有一些进展,如汪正华等[15]诱导表达了包涵体形式的mPG,再经变性、复性处理后测得酶活力为0.371 U/mL。刘英杰[28]构建的pHT43-pp/Bacillussubtilis168和pHT43-pp/BacillussubtilisDB403,在IPTG诱导下均可表达mPG,最终在浓缩发酵胞外液中获得约0.700 U/mL的酶活,其菌株产酶量为36.84 mg/mL。本文构建的表达菌株菌体上清液纯化后质量浓度为181.68 μg/mL,根据浓缩倍数计算出发酵液中酶量为47.19 mg/mL,比上述报道高10.39 mg/mL。尽管产酶量有一定的提高,但未实现胞外的可溶性表达。另外,本实验表达的酶仍需要通过外界酶进行加工,才可以释放有活性的PG。因此,建立高效表达PG的体系是实现PG大规模商业化应用的基础,也是今后的研究重点。
4 结论
本实验采用强组成型启动子P43与pro-mPG序列连接成功构建重组质粒pHT43:pro-mPG(P43),通过化学转化法成功获得BL21阳性工程菌,经PCR和基因测序验证正确。经Western blot验证和酶活性测定,证明菌体在发酵生长对数后期可通过群体感应自诱导表达目的蛋白,避免了IPTG的使用。通过对LB培养基中碳源、氮源的替换以及金属离子的添加,将酶活力由0.417 U/mL提高至0.761 U/mL。酶学性质研究表明,酶最适反应条件为45 ℃、pH 6.5。
