鸡肉中几种喹诺酮药物测定方法的优化
2021-02-25陈封政李书华温志国聂万丽
田 冲,刘 超,陈封政,李书华,温志国,李 骏,聂万丽※
(1.乐山师范学院 化学与资源环境学院,四川 乐山 614000;2.乐山市食品药品检验检测中心,四川 乐山 614000)
0 引言
喹诺酮药物是一种含有4-喹诺酮结构的人工合成药物。有着抗菌谱广、不良反应少、价格便宜等优点[1],因此广泛用于动物和人类的感染性疾病的治疗和预防[2]。对于禽类,喹诺酮药物主要用于由敏感菌导致的呼吸道、消化道、尿道感染和增加禽类饲料转化率[3]。在动物饲养中由于缺少相关专业知识或追求更高的经济效益导致喹诺酮药物在动物源性食品中残留的问题越来越严重[4]。通过食用喹诺酮残留超标的食品会造成耐药性和毒副作用,影响人体健康[5-6]。因此,世界各国都规定了喹诺酮药物在动物源性食品中的限量标准。
目前喹诺酮药物常用的检测方法有高效液相色谱法[7]、液相色谱-串联质谱法[8]、酶联免疫法[9]等,我国喹诺酮药物的检测方法主要有GB/T 20366—2006 和GB/T 21312—2007 等,这两种国标均使用了分离能力强、选择性高、灵敏度高的LC-MS/MS。GB/T 20366—2006 使用2%甲酸-乙腈作为提取液,提取液澄清,有利于净化;但标曲选择裸标,因基质效应会导致回收率偏低。GB/T 21312—2007 使用EDTAMcllvaine 缓冲液作为提取液,提取液浑浊,影响后续净化,使净化时间和成本增加。试验对两种国标进行比较并加以优化,以满足实验室检测需要。
1 材料与方法
1.1 材料
乙腈、冰乙酸、正己烷、甲酸、甲醇均为色谱纯;柠檬酸、磷酸氢二钠、氢氧化钠、乙二胺四乙酸二钠均为分析纯;HLB 固相萃取小柱(200 mg,6 mL)。
6 种喹诺酮标准品:洛美沙星(99.4%)Dr.Ehrenstorfer GmbH;诺氟沙星(97.2%)Dr.Ehren storfer GmbH;环丙沙星(99.3%)曼哈格;培弗沙星(99.7%)stanfordchem;氧氟沙星(99.0%)曼哈格;恩诺沙星(99.4%)曼哈格。
1.2 仪器与设备
高效液相色谱-串联质谱仪 DGU-20AAPI3200 AB SCIEX 公司;电子天平 FA124 上海舜宇恒平科学仪器有限公司;氮吹仪 HSC-B 天津市恒奥科技发展有限公司;固相萃取仪 VM24 天津艾杰尔科技有限公司;高速冷冻离心机 TGL-16M长沙湘智离心机仪器有限公司;水浴恒温振荡器SHA-B 常州冠军仪器制造有限公司;数控超声波清洗机 KQ700 昆山市超声仪器制造有限公司;超纯水机 UPT-Ⅱ-10T 四川优普超纯科技有限公司。
1.3 方法
1.3.1 样品前处理方法
a)按GB/T 20366—2006 进行提取和净化,其中将提取液用量改为10 mL,饱和乙腈正己烷用量改为15 mL;均质器均质1 min 改恒温震荡器震荡10 min,超声10 min;4 000 r/min 改为10 000 r/min;分液漏斗分液后旋蒸改为40 ℃氮吹,其余不变。
b)按GB/T 21312—2007 进行提取和净化,其中将提取液用量改为10 mL;10 000 r/min 涡旋1 min 改为恒温震荡器震荡10 min,超声10 min;过HLB 固相萃取柱前将提取液经过湿润滤纸过滤,其余不变。
1.3.2 标准溶液的配制
标准溶液的配制:取适量喹诺酮药物标准品用甲醇配制成1000μg/mL 标准储备液。将标准储备液用甲醇稀释至1μg/mL 的混合标准中间液。
混合标准工作液:分别移取适量混合标准中间液,甲醇稀释至系列浓度为5 ng/mL、10 ng/mL、20 ng/mL、40 ng/mL、80 ng/mL、100 ng/mL。
基质配标:取6 份5 g 阴性样品(精确到0.01 g)按1.3.1 方法制成空白基质溶液。分别取适量混合标准中间液,用空白基质稀释至系列浓度为5 ng/mL、10 ng/mL、20 ng/mL、40 ng/mL、80 ng/mL、100 ng/mL。
随行加标:取6份5 g阴性样品(精确到0.01 g),分别加入适量混合标准中间液,按1.3.1 方法提取净化。配制成系类浓度为5 ng/ml、10 ng/mL、20 ng/mL、40 ng/mL、80 ng/mL、100 ng/mL。
1.3.3 仪器条件
液相色谱条件:GB/T 20366—2006 色谱柱:Kromasil100-5-C18(3.9×150 mm)流动相:乙腈+0.2%甲酸溶液,梯度洗脱程序见表1。

表1 GB/T 20366—2006 梯度洗脱程序
流速:0.8 mL/min;进样量:20 μL。
GB/T 21312—2007色谱柱:C18柱(100*2.1 mm,1.7 μm)流动相:A[40+60 甲醇乙腈溶液];B[0.2%甲酸水溶液]梯度淋洗,梯度洗脱程序见表2。

表2 GB/T 21312—2007 梯度洗脱程序
流速0.2 mL/min;柱温40 ℃;进样量20 μL。
两种方法质谱条件均为:离子源ESI+;离子喷雾电压4500 v;雾化气氮气,30 ps;干燥气氮气,30 psi;离子化温度450 ℃;碰撞气氮气。
2 结果与分析
2.1 检出限和线性
分别按GB/T 20366—2006、GB/T 21312—2007规定,向3 个阴性样品中加入适量混合标准工作液,按1.3.1 处理,不断稀释,以三倍信噪比所对应浓度作为两种方法的检出限。详见表4。试验比较了GB/T 20366—2006、GB/T 21312—2007 两种方法的线性,结果见表5。

表3 喹诺酮药物的质谱分析参数
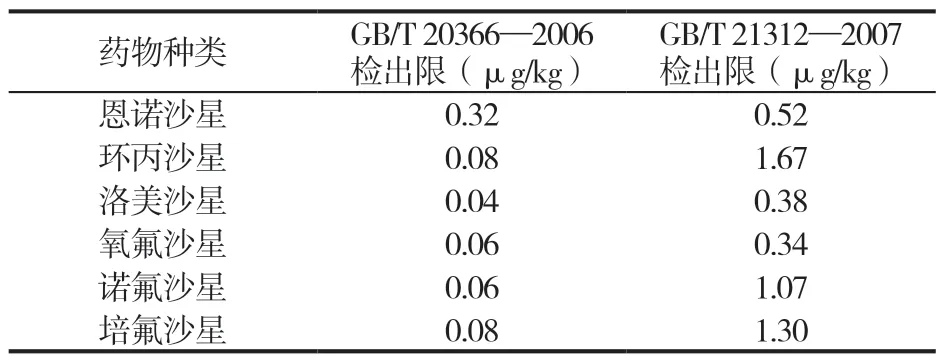
表4 两种方法中几种喹诺酮药物的检出限

表5 两种方法中几种喹诺酮药物的标准曲线
GB/T 20366—2006 几种喹诺酮药物检出限在0.04 μg/kg~0.32 μg/kg之间;GB/T 21312—2007几种喹诺酮药物检出限在0.34 μg/kg~1.67 μg/kg之间,表明GB/T 20366—2006 灵敏度高于GB/T 21312 —2007,但两种方法均满足检测要求。
由表5 可知,GB/T 20366—2006 中标曲采用裸标,标曲的相关系数R ≥0.998,有较好的线性相关,而GB/T 21312—2007 标准曲线由于采用随行加标,相关系数R 最小为0.991 最大为0.997 波动幅度较大,且线性不如GB/T 20366—2006。
2.2 精密度和加标回收率
于5 g(精确到0.01 g)阴性样品中分别加入10 ng、20 ng、40 ng 的混合标准溶液,按1.3.1 进行处理,每个浓度设置6 个平行样,由标准曲线计算对应回收率和相对标准偏差,详见表6 和表7。

表6 两种方法中几种喹诺酮药物的回收率(r=6)
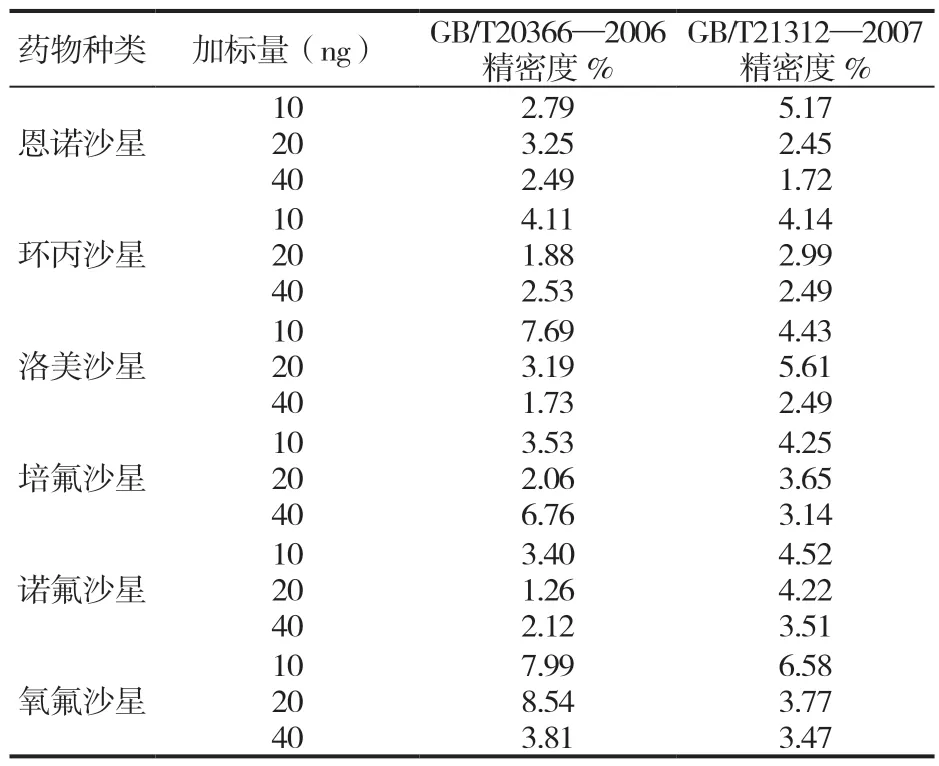
表7 两种方法中几种喹诺酮药物的精密度(r=6)
由表6 和表7 可知,GB/T 20366—2006 回收率为53.72%~71.30%,相对标准偏差为1.26%~ 7.69%。GB/T 21312—2007 回收率为84.37%~98.81%,相对标准偏差为1.72%~6.58%。GB/T 20366-2006 回收率较低,其中诺氟沙星回收率最低只有53.72%,可能的原因是GB/T 20366—2006 未考虑基质效应的影响,回收率较差。GB/T 21312—2007 回收率明显高于GB/T 20366—2006。两种国标精密度都满足检测要求。
2.3 基质效应
将基质配标的峰面积和裸标的峰面积通过公式ME(%)=(A/B)*100%[10]计算:
A 为裸标的峰面积;B 为基质配标的峰面积。当ME <100%时,基质对化合物产生抑制作用;当ME >100%时,基质对化合物产生增强作用;当ME=100%时,无基质效应。
试验比较了两种方法在浓度分别为5 ng/mL、20ng/mL、40 ng/mL、80 ng/mL 的基质效应。结果见表8。
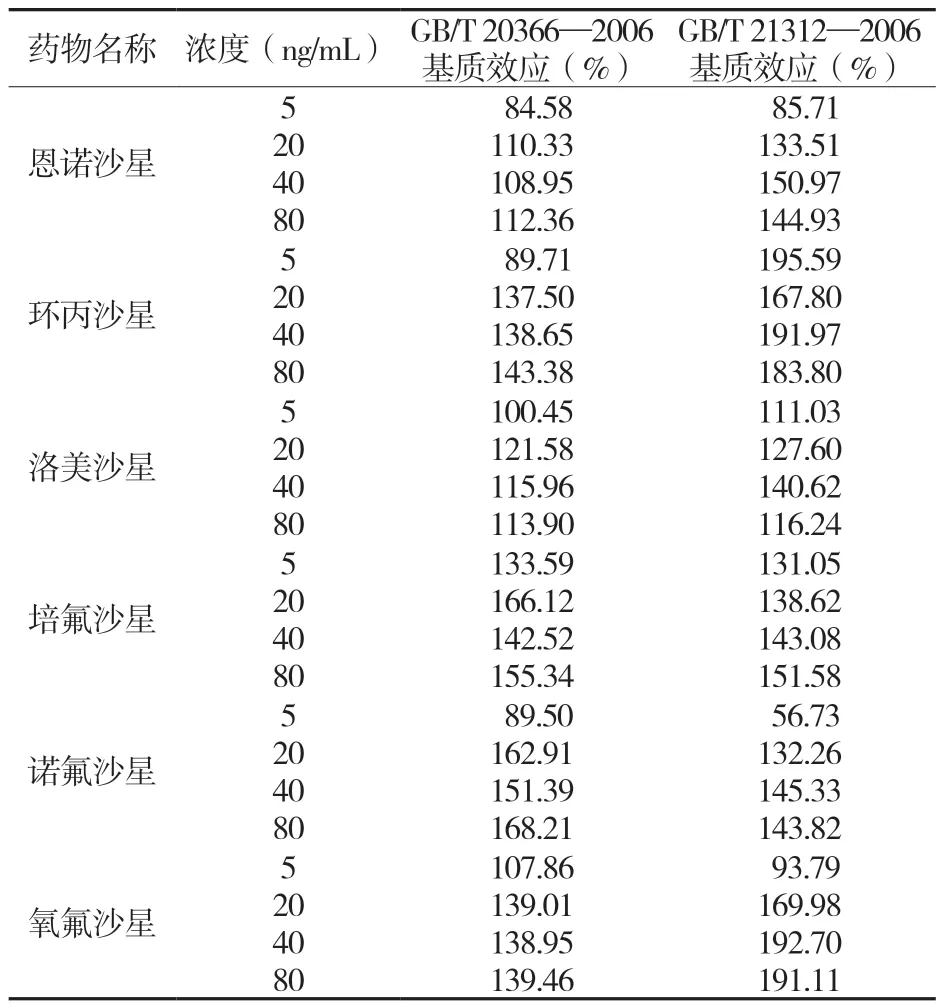
表8 两种方法中几种喹诺酮药物的基质效应
由表8 可知。GB/T 20366—2006、GB/T 21312—2007 均有明显的基质效应,且不同化合物、同一不同浓度的基质效应影响不一;整体来说,GB/T 20366—2006 的基质效应较GB/T 21312—2007 小。由此可见,基质效应对LC-MS/MS 检测喹诺酮类药物的响应造成了不可忽视的影响,由于GB/T 21312—2007 采用随行加标,消除了基质效应,因此回收率较高;而GB/T 20366—2006 采用裸标,忽视了基质效应,因此回收率较低。
2.4 方法的优化
鸡肉中基质较为复杂,基质效应较大,GB/T 20366—2006 使用裸标导致回收率偏低,试验改为基质配标或随行加标,考察是否能抵消或者消除基质效应。GB/T 21312—2007 使用随性加标的方式,线性较差,试验考察基质配标对回收率及线性的影响;EDTA-Mcllvaine 缓冲液提取会提取出鸡肉组织中的水溶性杂质和部分蛋白质导致过前处理小柱时堵塞,使用滤纸过滤后再过前处理小柱能有效避免前处理小柱堵塞。结果见表9、表10。

表9 优化后两种方法中几种喹诺酮药物的回收率(r=6)

表10 优化后两种方法中几种喹诺酮药物的精密度(r=6)
由表9、表10 可知,优化后,GB/T 20366—2006采用基质配标回收率在68.17%~83.77%之间,相对标准偏差在1.38%~5.47%之间;采用随行加标,回收率在79.75%~92.88%之间,相对标准偏差在1.22%~5.41%之间,由此可见,采用基质配标和随行加标均能提高回收率,随行加标不仅消除了基质效应,还考虑到实验的操作误差,因此加标回收率更好,因此,GB/T 20366—2006 采用随行加标作为方法的标准曲线。GB/T 21312—2007采用基质配标线性较原标准的线性好,但是回收率在70.38%~89.27%之间,不及原标准,因此GB/T 21312—2007 采用原标准的标准曲线。
3 结论
试验通过检出限、线性、回收率、重复性、基质效应对两种方法进行比较。结果显示,由于鸡肉基质复杂,两种国标均有着不可忽视的基质效应。GB/T 20366—2006 提取和净化时间明显较短,操作更加便捷,有着更高的灵敏度和精密度,但回收率偏低;GB/T 21312—2007 有着更高的回收率。GB/T 20366—2006 在改进之后回收率显著提高,检测结果更加准确。GB/T 21312—2007 改进之后,使用滤纸过滤后再过前处理小柱能有效避免前处理小柱堵塞,节约检测时间,提高检测效率。两种方法在优化之后都能满足实验室检测需要,因此在应用中可以根据具体的条件选择不同优化之后的方法来获取更好的实验结果。
