盐酸苯达莫司汀原料药含量测定方法的比较
2021-02-23沈红梅
沈红梅
(四川科伦药物研究院有限公司,四川 成都 611130)
盐酸苯达莫司汀是一种烷化剂类抗肿瘤药,用于恶性淋巴瘤(何杰金氏病HD和非何杰金氏淋巴瘤NHL)、慢性淋巴细胞性白血病(CLL)、多发性骨髓瘤(MM)和乳腺癌的治疗[1-2]。由于盐酸苯达莫司汀对多种血液系统肿瘤和实体瘤具有良好疗效,且毒副作用低[3-4],目前已批准用于多种癌症的治疗[5-6]。
为了确保该原料药的质量,探索盐酸苯达莫司汀原料药含量测定方法的多元化,本研究参考相关文献[7],首先,建立了高效液相色谱法测定盐酸苯达莫司汀的含量,考察线性关系、稳定性、精密度等;然后,根据盐酸苯达莫司汀的性质,建立了非水滴定法测定其含量,进一步通过实验比较两种测定方法的差异性。
1 仪器与试剂
1.1 仪 器
Aglient 1100液相色谱仪,二极管阵列SPD-10MAVP(DAD)检测器及Aglient 1100紫外检测器,定量进样阀,Aglient 1100色谱工作站;Shimadzu VP-ODS 150 L×4.6 mm 5 μm色谱柱。
1.2 试 药
盐酸苯达莫司汀(批号: 20190901、20190902、20190903)、盐酸苯达莫司汀对照品(批号:20190701s)均来自于四川科伦药物研究院有限公司;乙腈为色谱纯,三乙胺为分析纯,水为重蒸馏水。
2 实验方法
2.1 HPLC法测定
2.1.1 色谱条件及仪器
仪器为Aglient 1100,检测器为Aglient 1100 UV检测器,流动相为乙腈-0.25%三乙胺溶液(用H3PO4调pH至2.4±0.2)(体积比为30:70),色谱柱为Shimadzu VP-ODS 150 L×4.6 mm 5 μm,流速为1.0 mL·min-1,UV检测波长为233 nm,色谱工作站为Aglient 1100色谱工作站(AGLIENT公司);进样量20 μL。
2.1.2 测定方法
精密称取经五氧化二磷干燥过夜的苯达莫司汀对照品约20 mg,置100 mL量瓶中,加冰浴中放置后的流动相振摇使溶解并稀释至刻度,摇匀,精密量取10 mL置100 mL量瓶中,加上述溶液稀释至刻度,摇匀,作为对照品溶液。
精密称取本品约20 mg,置100 mL量瓶中,加冰浴中放置后的流动相振摇使溶解并稀释至刻度,摇匀,再精密量取10 mL置100 mL量瓶中,加上述溶液稀释至刻度,摇匀,作为供试品溶液。
分别取上述对照品溶液和供试品溶液20 μL注入液相色谱系统,记录色谱图。按外标法以峰面积计算,即得。
2.1.3 线性范围考察
精密称取盐酸苯达莫司汀对照品25.71 mg置100 mL量瓶中,加入流动相溶解,稀释至刻度,得1号溶液(257.1 μg/mL)。精密吸取得1号溶液各10 mL、8 mL、6 mL、5 mL、4 mL、3 mL、2 mL于50 mL量瓶中,稀释至刻度,得2号(51.420 μg/mL)、3号(41.136 μg/mL)、4号(30.852 μg/mL)、5号(25.710 μg/mL)、6号(20.568 μg/mL)、7号(15.426 μg/mL)、8号(10.284 μg/mL)溶液。分别取上述溶液按含量测定法测定,结果见表1。

表1 线性测定结果
从表1可以看出,苯达莫司汀浓度在10.284~41.1 μg/mL线性范围内峰面积与浓度呈良好的线性关系,线性方程为A=86.6862C+13.3345(其中:A-HPLC图中主峰峰面积,C-样品溶液浓度),相关系数为0.99992。
2.1.4 稳定性考察
按“1.2”法制得供试品溶液,分别于0、1、2、3、4 h测定,试验结果见表2。由表2可知,供试品溶液在1 h内主峰面积下降1%,故含量测定在1 h内测定。

表2 稳定性试验结果
2.1.5 精密度考察
为考察随机变动因素对精密度的影响,由不同分析人员在不同日期各取同一批样品适量(批号:20190701)9份,精密称定,加冰浴中放置的流动相使溶解,并配制成每1 mL约含盐酸苯达莫斯汀16~24 μg的溶液,在不同仪器上照含量测定项下,依法进行测定。试验结果见表3。由表3可知,精密度试验表明,精密度好,符合高效液相色谱法含量测定要求。该方法精密度高,能达到仪器分析要求。
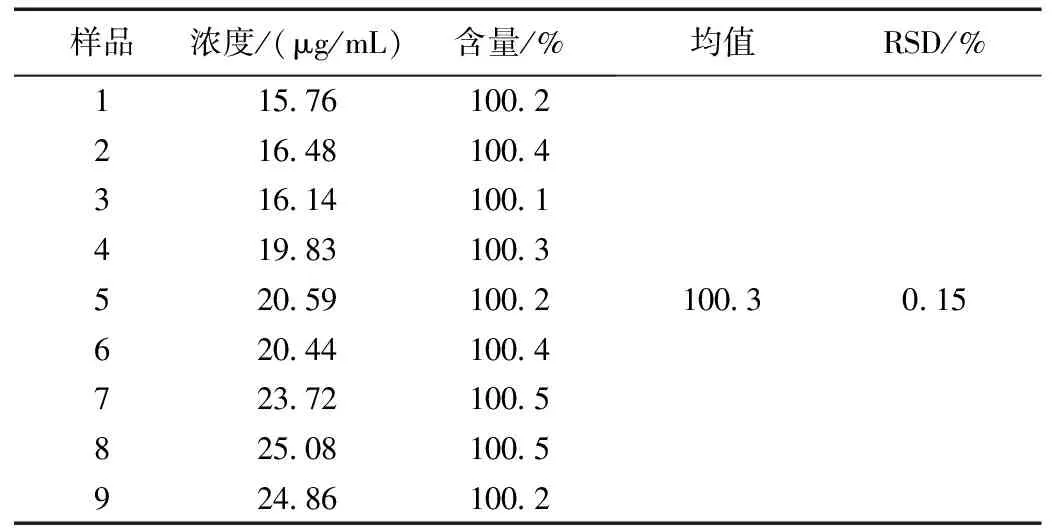
表3 精密度试验结果
2.1.6 含量测定
精密称取5种不同批号盐酸苯达莫司汀约20 mg,分别置20 mL量瓶中,稀释至刻度;分别取10 μL注入高效液相色谱仪,并记录峰面积,用外标法计算盐酸苯达莫司汀的含量,结果见表4。

表4 含量测定结果
2.2 非水溶液滴定测定
2.2.1 测定原理
根据本品结构特点及性质,可采用非水滴定法测定其含量。其机理如下:
2C16H21Cl2N3O2·HCl+Hg(Ac)2→C16H21Cl2N3O2H+·Ac-+HgCl2
2.2.2 测定方法
精密称定本品30 mg,加冰乙酸5 mL使溶解后,加醋酐25 mL与醋酸汞试液5 mL,按照电位滴定法(中国药典2020年版二部附录Ⅶ A),用高氯酸滴定液(0.1 mol/L)滴定,并将滴定结果用空白试验校正。每1 mL高氯酸滴定液(0.1 mol/L)相当于39.47 mg的C16H21Cl2N3O2·HCl,滴定数据见表5。

表5 电位滴定结果
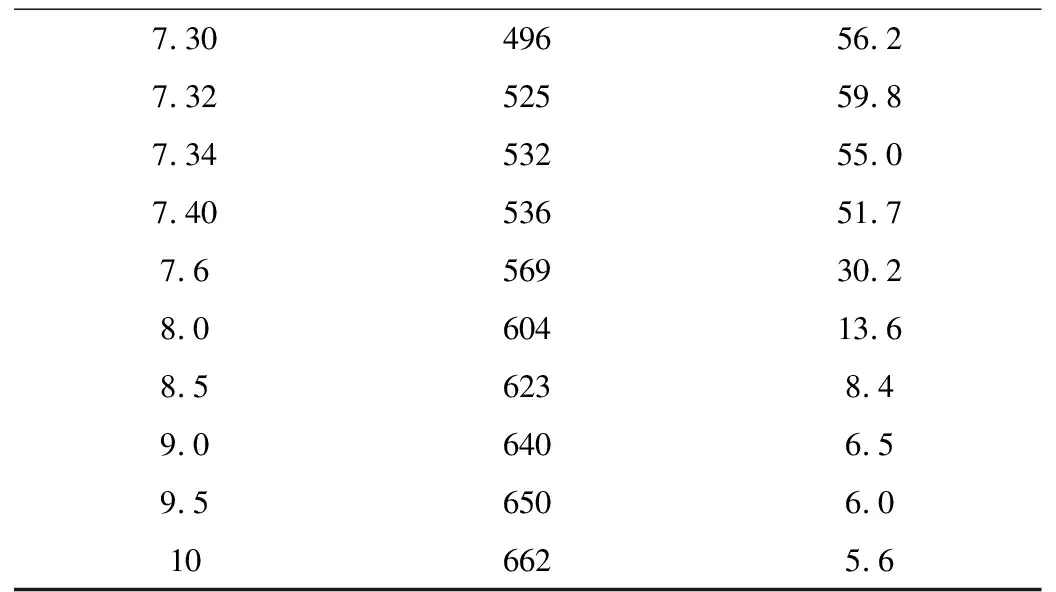
续表5
2.2.3 精密度
精密取本品(批号:20190701)9份,按上述含量测定方法,按高、中、低浓度测定本品,试验结果见表6。由表6可知,该方法精密度高,重现性好,达到容量分析的要求。
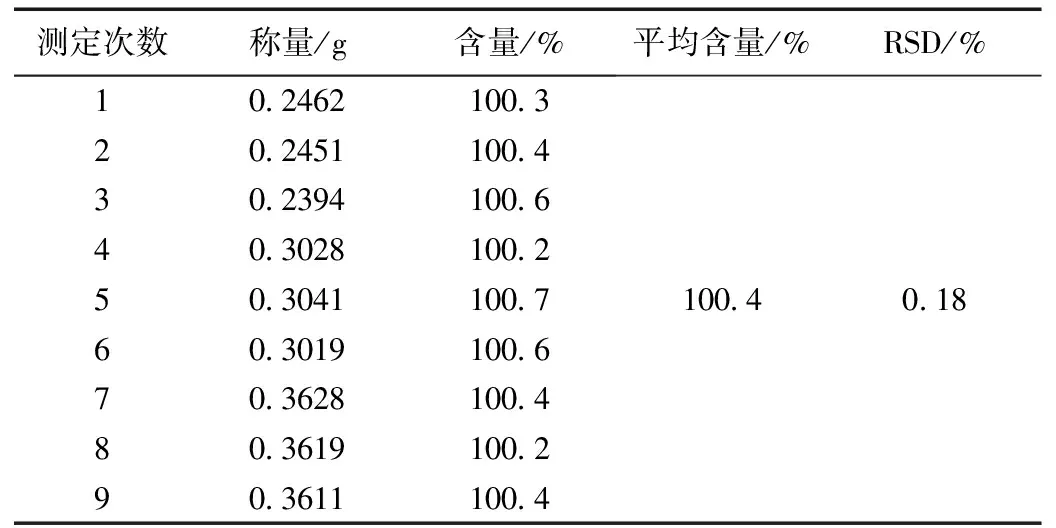
表6 精密度测定结果
2.2.4 含量测定
精密称取5批盐酸苯达莫司汀样品30 mg,按照“2.2.2”的方法测定含量。结果见表7。由表可知,五批样品含量均大于99.5%。该方法可靠。

表7 非水滴定含量测定结果
2.3 两种测定方法结果的比较
称取盐酸苯达莫司汀原料药,分别采用HPLC法和非水滴定法测定其含量,结果见表8,结果通过t检验分析[P=0.18(>0.05)],表明两种方法的测定结果 无显著性差异(α=0.05)。
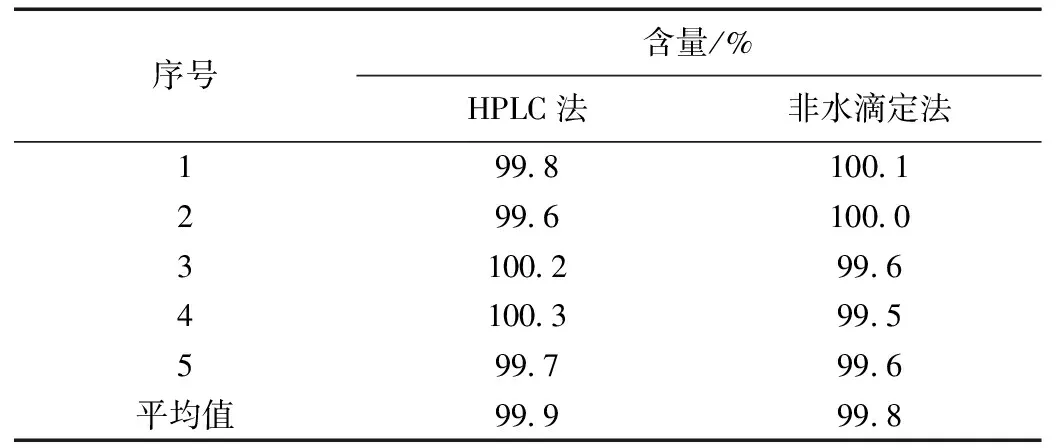
表8 两种方法测定含量的结果
3 讨 论
(1)在选择流动相时,根据文献色谱条件[7],主峰严重拖尾,换用不同的缓冲盐拖尾现象得不到明显改善,后换用乙腈-0.25%三乙胺溶液为流动相,拖尾现象得到明显改善,因此选用的流动相是乙腈-0.25%三乙胺溶液(用H3PO4调pH至2.4)(体积比为30:70)。
(2)原料药盐酸苯达莫司汀的含量测定采用非水滴定法与HPLC法结果基本一致。由于非水滴定快速,故首选容量分析法测定本品含量。
4 结 论
采用非水滴定法测定盐酸苯达莫司汀的含量,具有快速、准确等优点,可作为盐酸苯达莫司汀含量测定方法的参考和对比研究。
