Pancreatic β cell regeneration induced by clinical and preclinical agents
2021-02-23KangLiWangMingTaoTianJiaoWeiRuiWei
Kang-Li Wang, Ming Tao, Tian-Jiao Wei, Rui Wei
Kang-Li Wang, Tian-Jiao Wei, Rui Wei, Department of Endocrinology and Metabolism, Peking University Third Hospital, Beijing 100191, China
Ming Tao, Department of General Surgery, Peking University Third Hospital, Beijing 100191, China
Abstract Diabetes, one of the most common chronic diseases in the modern world, has pancreatic β cell deficiency as a major part of its pathophysiological mechanism.Pancreatic regeneration is a potential therapeutic strategy for the recovery of β cell loss.However, endocrine islets have limited regenerative capacity, especially in adult humans.Almost all hypoglycemic drugs can protect β cells by inhibiting β cell apoptosis and dedifferentiation via correction of hyperglycemia and amelioration of the consequent inflammation and oxidative stress.Several agents, including glucagon-like peptide-1 and γ-aminobutyric acid, have been shown to promote β cell proliferation, which is considered the main source of the regenerated β cells in adult rodents, but with less clarity in humans.Pancreatic progenitor cells might exist and be activated under particular circumstances.Artemisinins and γ-aminobutyric acid can induce α-to-β cell conversion, although some disputes exist.Intestinal endocrine progenitors can transdeterminate into insulin-producing cells in the gut after FoxO1 deletion, and pharmacological research into FoxO1 inhibition is ongoing.Other cells, including pancreatic acinar cells, can transdifferentiate into β cells, and clinical and preclinical strategies are currently underway.In this review, we summarize the clinical and preclinical agents used in different approaches for β cell regeneration and make some suggestions regarding future perspectives for clinical application.
Key Words: β cell regeneration; β cell dedifferentiation; Cell proliferation; Pancreatic progenitors; α-to-β cell transdifferentiation; Enteroendocrine progenitor cells
INTRODUCTION
Diabetes is a critical global health concern due to its high prevalence[1], as well as related disability and mortality[2].Pancreatic β cell deficiency is a major component of the pathophysiological mechanism[3].Substantial β cell loss results in permanent endocrine deficiency and irreversible diabetes.Pancreatic regeneration is a potential therapeutic strategy for β cell recovery.However, the endocrine pancreas (islet) has limited regenerative capacity, especially in adults[4].Therefore, strategies for promoting β cell regeneration have profound implications for the treatment of diabetes, especially for type 1 diabetes (T1D) and late-stage type 2 diabetes (T2D) with substantial β cell loss.
There are two ways to regenerate pancreatic β cells.The first is to avoid β cell loss, including inhibiting β cell apoptosis/necrosis and dedifferentiation.The second is to promote newborn, including endogenous regeneration (stimulating β cell proliferation, inducing transdifferentiation and transdetermination of other cells to β cells, reactivating pancreatic endocrine progenitors, and facilitating differentiation into β cellsin vivo) and exogenous supplementation (promoting the differentiation of stem cells into β cells, inducing the reprogramming of mature cells into β cellsin vitro, and then transplanting the obtained cells to patients with diabetes).There is a long history of investigations regarding pancreatic regeneration that dates back to nearly a century.Under some physiological conditions (e.g., during pregnancy, in obesity, and under insulin resistance conditions), islet adaption and increased β cell mass occur in both animal models and humans[5-8].Recently, the emerging technologies have supplied more evidence for β cell regeneration.Single-cell RNA sequencing data have showed that human islets contain four distinct subtypes of β cells[9], as well as potentially intermediate stages[10], suggesting that β cells can adapt, transdifferentiate, or undergo neogenesis.Investigations regarding physiological regeneration may provide information for drug development.Several strategies have been used to promote β cell regeneration, including pancreatectomy, partial duct ligation, and chemical-induced massive β cell loss[11-15].The molecular pathways that drive increases in β cell mass have been widely studied.Thousands of materials have been investigated, and hundreds have been proved to be functional in the process of β cell regeneration, but only a small proportion are clinical, pre-clinical, or clinical potential drugs.
CORRECTION OF METABOLIC DISORDER AND INHIBITION OF APOPTOSIS
The main clinical manifestation of diabetes is hyperglycemia, usually accompanied by dyslipidemia.Glucotoxicity, lipotoxicity, and the subsequent inflammation and oxidative stress result in β cell dysfunction and death, which further exacerbate hyperglycemia and create a vicious circle.Therefore, antidiabetic drugs can lower glycaemia and disturb the circle, and thus, inhibit functional β cell loss[16-18].However, the gradual loss of β cell function cannot be reversed using recently developed therapies.In some cases, earlier and longer use of the hypoglycemic drug has better protective effects on β cells[19].Considering the wide existence of autoimmune damage in T1D and islet microenvironment inflammation in T2D, some immunomodulation therapies can protect β cells[20].For instance, the activation of the nuclear receptor LRH-1/NR5A2 induces immune self-tolerance and increases β cell survival[21].CD3 monoclonal antibody (mAb), which suppresses the immune system, has significant albeit transient preservation capabilities of β cell function, resulting in a decrease in insulin requirements for at least 24 mo in patients with recent-onset T1D, and delays clinical symptoms by approximately 2 years in high-risk relatives of patients with T1D[22].γ-aminobutyric acid (GABA) acts as an immunosuppressive regulator in T1D by mediating cytokine secretion from human peripheral blood mononuclear cells and CD4+T cells[23].Notably, the antidiabetic drug glucagon-like peptide-1 (GLP-1) analogue directly modulates innate immunity-mediated inflammation in patients with T2D[24].In summary, the hypoglycemic drugs and immunomodulation therapies can protect β cells,viadelay or prevention of β cell loss, by correcting metabolic disorders and improving the microenvironment.
Except for the indirect effects, several antidiabetic drugs have direct protective effects on β cells by inhibiting stress-induced cell death.The most remarkable drug is GLP-1.Native GLP-1 and GLP-1 analogs (e.g., liraglutide, exenatide, and lixisenatide) exert antiapoptotic effects on human and rodent β cells through AMPK/mTOR and PI3k/Akt signaling pathways, as previously summarized[25].In addition, GLP-1 prevents pancreatic β cell death by increasing autophagic flux and restoring lysosomal function[26].The classic hypoglycemic drug metformin decreases human islet apoptosis, increases insulin content, and increases the number and density of mature insulin granules, which is mediated by a reduction in oxidative stress[27].Other clinical drugs, such as angiotensin-converting enzyme inhibitors, protect human islets from glucotoxicity by inhibiting oxidative stress[28].In summary, the direct antiapoptotic effect, together with indirect metabolic improvement and amelioration of inflammation, attenuates β cell loss.
INHIBITION OF β CELL DEDIFFERENTIATION
Cell dedifferentiation is considered a mechanism of diabetic β cell failure[29].Following physiological or pathological stress, β cells lose fully mature healthy characteristics and revert to progenitor-like cells, with changes in gene expression patterns (upregulation of genes that are typically expressed in embryonic endocrine progenitors, and downregulation of genes that are expressed in mature β cells) and in structural and functional elements (loss of mature secretory granule).The Accili group and other groups extend this concept to humans and observe chromogranin A/synaptophysin+and hormone-endocrine cells in patients with T2D and T1D[30-32].The Accili group concludes that an approximately 30% deficit of β cells in T2D is not due to death but instead to dedifferentiation or transdifferentiation of β cells to other islet types[30].In contrast, the Butler group finds that the chromogranin+hormone-islet cells accounts for no more than a 2% deficit of β cells in T2D[33].Single-cell transcriptomics in islets from young children reveal that multiple α cell signature genes are preferentially observed in juvenile β cells, suggesting the immature state.This pattern is also observed in T2D donors, indicating a dedifferentiation process[34].Nevertheless, there is no consistent conclusion regarding cell dedifferentiation in humans due to the incapability of lineage tracing.
This concept of dedifferentiation is of great significance for clinical treatments.In contrast to cell death, β cells do not disappear; they exist but lose their characteristics, implying that the dedifferentiated cells can quickly restore the functional β cell mass.For instance, dysfunctional β cells can recover in patients with T2D with proper management, such as diet, exercise, or intensive insulin therapy[35,36].Except for the antidiabetic drugs and the diabetes management, other drugs also own the ability of inhibition of β cell dedifferentiation.Salsalate, an anti-inflammatory drug with antidiabetic properties, diminishes β cell dedifferentiation by inhibiting the Notch1 pathway[37].Renin–angiotensin system inhibitors, either angiotensin II type 1 receptor blockers or angiotensin-converting enzyme inhibitors, efficiently reverse the dedifferentiated status of β cellsviainhibition of NF-κb signaling[38].It should be noted that continued intervention may be required to alter the progressive loss of β cell function.As is shown by a recent report, liraglutide plus metformin improves β cell function during the treatment period, but the effects disappears after the treatment is stopped, in adults with impaired glucose tolerance or newly-diagnosed T2D[39].
STIMULATION OF β CELL PROLIFERATION
Cell proliferation is considered the main source of regenerated β cells in adult rodents[40].The proliferative rate of β cells is notably high in fetal and neonatal rodents but declines rapidly with age[41].A large number of growth factors and mitogenic agents have been shown to promote β cell proliferation in animal models.These include hepatocyte growth factor, GLP-1, insulin-like growth factors, epidermal growth factors, and others[42].However, these agents have generally failed to promote the significant proliferation of human β cells, possibly due to molecular, structural, and functional differences, as well as developmental disparities between mouse and human islets[43-45].In fact, the normal turnover of human β cells is considerably lower than that of mice[4,46], and β cells adapt to stressors (such as pregnancy or obesity) in completely different ways[47].Therefore, further consideration should be given to the validity of the mouse model to draw conclusions in humans.Thanks to highthroughput screening technology and the availability of human islets, an increasing amount of information regarding human β cells is becoming available.Tyrosineregulated kinases Dyrk1a and Nfat manipulate human β cell proliferation[48].Hepatocyte-derived secretory SerpinB1 and its partial mimic GW311616A enhance β cell proliferation by inhibiting elastase activity and activating key proteins in the growth factor signaling pathway[49].GABA promotes β cell replication in grafted human islets by activating a calcium-dependent signaling pathway and the downstream PI3K/Akt and CREB-IRS2 signaling pathways[50].The antidiabetic drug GLP-1 analog exendin-4 stimulates human β cell proliferation in juvenile, but not adult, islets and requires calcineurin/Nfat signaling[51].However, human β cells appear to resist proliferation, and their proliferation response to mitogenic stimuli is limited at an approximately 0.3%-0.5% increase compared to the basal proliferation index (approximately 0.1%-0.2%)[46].Notably, combining of the Dyrk1a inhibitor with the GLP-1 receptor agonist[52], or combined inhibition of Dyrk1a, SMAD, and Trithorax pathways[53]induces a synergistic increase in human β cell replication (5%-8%).Nevertheless, the approach of stimulating β cell self-replication is not applicable for treating patients with complete or near-complete absence of β cells.
PROMOTION OF STEM CELL DIFFERENTIATION
Stem cells, possessing abilities of self-renewal and multilineage differentiation, are the ideal source for cell replacement therapy.There are two methods of stem cell-based β cell regeneration.One is to promote stem cell differentiation into β cellsin vitro.The other is to activate pancreatic progenitors and promote them to differentiate into β cellsin situ.Pluripotent stem cells, including embryonic stem cells (ESCs) and induced pluripotent stem cells, which have unlimited self-renewal abilities, are appealing seed cells for β cell regeneration.A stepwise protocol was established to guide cell differentiation through four successive stages (definitive endoderm, pancreatic epithelium, endocrine progenitors, and β-like cells)[54].The stepwise protocol replicates the signaling events that control β cell formation during human pancreas development.At present, most differentiation protocols are based on this protocol.After decades of work, optimized cocktails of cytokines and chemicals have yielded cells with remarkable transcriptional, morphological, and functional resemblance to bona fide β cells[55,56].More recent studies have defined conditions that greatly improve the functional maturation of β cells, achieving first- and second-phase insulin secretion[57,58].Notably, diabetic patient-specific β cells can be derived from induced pluripotent stem cells or nuclear transfer ESCs, which can avoid immune rejection[59,60].Except for the stem cell-derived β cells, stem cell-derived pancreatic endoderm cells or pancreatic progenitors can also be the source of cell replacement for diabetes treatment[61].Transplantation of these progenitors has led to further functional maturationin vivoand rescue of experimental diabetes in mice[62].Other stem cells, including mesenchymal stem cells (MSCs), can also differentiate into β cellsviathe stepwise induction protocol[63].Interestingly, MSCs (unlike MSC-derived β-like cells) protect β cells and promote islet regeneration by secreting numerous immunomodulatory and tissue regenerative factors[64], thereby inhibiting inflammation in the islet microenvironment and promoting microcirculation.At present, ESC-derived pancreatic progenitors and MSCs are already undergoing clinical trials.Duringin vitrostepwise differentiation, different protocols are used.In each step, many cytokines and chemicals are used to induce stem cell differentiation toward β cells.It is worth mentioning that the antidiabetic drug GLP-1 promotes ESCs (including mouse and human) and other stem cells (e.g., pancreatic progenitors) to differentiate into β cells[65-68].Other clinical agents, including ascorbic acid, zinc sulfate, and N-acetyl cysteine, are also used for β cell differentiation[56,58].However, the clinical agents cannot work alone and must interact with other agents.
There is a long-standing hypothesis that pancreatic stem or progenitor cells exist in the adult animal or even human pancreas.In most circumstances, the pancreatic duct serves as a pool for progenitors of both endocrine and exocrine cells after birth and into adulthood[69].The early studies have showed that after pancreatectomy or pancreatic duct ligation, a rare population of Ngn3+endocrine progenitors appears in ductal structures in mouse models[14,15].A recent study also shows that Ngn3+cells are around islets and ducts in experimental models of α-to-β cell transdifferentiation[70].The notion of pancreatic duct-derived neogenesis has been confirmed by using lineage-tracing experiments in mice.However, lineage tracing cannot be conducted in the human pancreas, and there are some conflicting findings, but more evidence supports this notion than not.In pregnant, obese, insulin-resistant, and diabetic humans, the number of single/small clusters of insulin+cells and bihormoneexpressing cells, as well as the proportion of insulin+cells, within ducts has been observed to increase, suggesting that neogenesis may be the main mechanism in adult humans[7,8,71].A very recent study conducts single-cell RNA sequencing and confirms the existence of multipotent progenitor-like cells within the pancreatic ducts of the human pancreas in patients with T1D and T2D, regardless of the duration of the disease[72].Nevertheless, investigations into the specific markers that characterize the progenitors are still in process.Carbonic anhydrase II[73], Hnf1β, Sox9[74], stage-specific embryonic antigen 4[75], and activin-like kinase 3[72]are suggested as progenitor markers but the results are inconsistent[76].Notably, except for the ductal tree, the progenitors might also exist in intraislets.The recent identification of a “virgin β cell subpopulation”, a urocortin 3-, MafA-, and insulin+subpopulation in the periphery of the islet, adds to the potential list of progenitors that can contribute toward a functional β cell pool[77].A very recent study finds that the adult mouse islets contain a population of protein C receptor-positive endocrine progenitors, which undergo clonal expansion and generate all four endocrine cell types during adult homeostasis[78].For clinical drugs, GLP-1/exendin-4 has been reported to facilitate β cell neogenesis from duct cells in streptozocin-induced T1D rats and in cultured human ducts[79].The dipeptidyl peptidase 4 (DPP4) inhibitor (which inhibits GLP-1 degradation) vildagliptin promotes β cell neogenesis in streptozotocin-induced diabetic rodents[80,81].Interestingly, a novel diet therapy with a 4-d fasting-mimicking diet induces a stepwise expression of Sox17 and Pdx1 (resembling that observed during pancreatic development), followed by the Ngn3-driven neogenesis of β cells, and restores insulin secretion and glucose homeostasis in both T1D and T2D mice.In human T1D pancreatic islets, fasting conditions also activate pancreatic progenitors and promote insulin production[82].
INDUCTION OF CELL TRANSDIFFERENTIATION AND TRANSDETERMINATION
Cell transdifferentiation, also known as lineage reprogramming, can also be used for β cell regeneration.Distantly related cells (e.g., fibroblasts and keratinocytes) and developmentally related cells (e.g., liver, gastrointestinal, and pancreatic exocrine cells) can be converted into functional β cells (summarized in our previous review[83]).Endogenous α and δ cells are attractive sources for β cell reprogramming due to their same developmental transcriptional mechanisms, similar epigenetic landscape, and distinctive location.Under a condition of more than 99% β cell loss, slow but significant recovery of β cell mass occurs in mice.Lineage-tracing studies suggest that the new insulin-producing cells arise from the conversion of pancreatic α cells (in adults) or δ-cells (before puberty)[12,13].The molecular mechanism of this conversion between islet cell phenotypes is currently unknown.The genetic deletion of Arx (a master regulator of α cell development, and a key transcription factor for maintenance of the α cell identity) or forced expression of Pax4 (a master regulator of β cell development, and a key transcription factor for maintenance of the β cell identity) converts α cells to β cells in mouse models[84,85].Forced expression of Pdx1 and MafA induces the transdifferentiation of human α cells to β cells[86].The ability of cell transdifferentiation is confirmed by lineage-tracing in mice, and preclinical strategies are underway.GABA is an inducer of α-to-β cell conversion in mice and human islets, which is useful information for clinical trials[87].Artemisinins, which have already been used for malaria treatment clinically, improve glucose-stimulated insulin release, change gene profiles of human islets, and induce α-to-β transdifferentiation[88].These data provide a possible unprecedented β cell regeneration strategy using a known and approved therapeutic agent.However some paradoxical effects of artemisinin on β cell regeneration warrant further verification[89].The widely used hypoglycemic drug GLP-1 has showed some interesting results.An early report has found that 9-wk treatment of liraglutide increases β cell number and insulin content and secretion, while it decreases α cell number in ZDF rats[90].A recent study by Leeet al[91]finds that recombinant adenovirus expressing GLP-1 can promote α-to-β cell conversion in streptozotocin-induced T1D mice, which is proved by using lineage-tracing technology[91].The determination of whether the currently marketed GLP-1 drugs (including GLP-1 analogs and DPP4 inhibitor) and other drugs that can increase GLP-1 levels can induce phenotype conversion from α cells to β cells is of considerable interest.In fact, we prove that glucagon receptor mAb, which increases GLP-1 secretion in the islets and intestines[92,93], can induce α-to-β cell conversion in T1D mice[93].Our most recent study reveals that dapagliflozin, a sodium-glucose cotransporter type 2 inhibitor, also induces α-to-β cell conversion in T2D mice[94].Lee’s study concludes that fibroblast growth factor 21 (FGF21) participates in GLP-1-induced α-to-β cell conversion[91], suggesting that FGF21, which is undergoing clinical trials[95], might also possess this ability.In contrast to α-to-β cell conversion, δ cellderived β cell regeneration is faster and more efficient, always leading to diabetes recovery, but only occurs before puberty[13].Further study shows that instead of direct conversion, δ-cells dedifferentiate to a progenitor stage, reenter the cell cycle, and recapitulate embryonic development to become insulin producers[13].This juvenile adaptability relies, at least in part, upon the combined action of FoxO1 and downstream effectors.The juvenile mechanism can be somewhat mimicked with the pharmacological inhibition of FoxO1 after injury, which promotes the δ-to-β conversion in adulthood[13].
Acinar cells comprise the most abundant pancreatic cell type, and therefore, constitute an attractive source for β cell reprogramming.A combination of Ngn3, Pdx1, and MafA (three developmental regulators of β cells) efficiently converts pancreatic acinar cells into β-like cells after delivery into the adult mouse pancreas using adenoviral vectors[96].The lineage-reprogrammed cells achieve long-term stability and undergo epigenetic, transcriptional, anatomical, and functional development toward a β cell phenotype, as well as acquire the ability to reverse diabetes[97].A transient cytokine mixture of epidermal growth factor and ciliary neurotrophic factor activates Stat3 signaling, leads toin vivoconversion of acinar-to-β cells, and reverses alloxan-induced diabetes in mice[98].Without any genetic manipulation, bone morphogenetic protein 7 induces the conversion of adult human nonendocrine pancreatic tissue into endocrine cell types[99].However, the insulinexpressing cells arise mainly from extrainsular progenitors rather than the mature exocrine cells[99].It appears that there is not an easy way to induce phenotype conversion of mature acinar cells to endocrine cellsviaclinic-related strategies.
Enteroendocrine progenitors are ideal cell sources for insulin production.Enteroendocrine progenitors express Ngn3, the same marker as pancreatic endocrine progenitors, and continually arise from gut stem cells as well as contribute to the repopulation of the high-turnover enteroendocrine population.Conditional deletion of FoxO1 from Ngn3+intestinal endocrine progenitors leads to the formation of insulinproducing cells in the gut (namely, transdetermination)[100].Pharmacological or RNA interference-based FoxO1 inhibition in the gut might also achieve this goal.The inhibitors of FoxO1 have been studied extensively, but to date, their application in the management of metabolic disorders is limited[101].Oral administration of the FoxO1 inhibitor AS1842856 to diabeticdb/dbmice leads to a drastic decrease in fasting plasma glucose levels[102], which is suggestive of clinical potential.We have previously showed that FoxO1 inhibition by an inhibitor or siRNA promotes human ESCs to differentiate into β cells[103].Based on these results, we infer that the FoxO1 inhibitor might promote the production of β cells from intestinal endocrine progenitors.Another target for intestine-to-β cell conversion is GLP-1.Intraintestinal injection of a recombinant adenovirus constitutively expressing GLP-1 produces insulin-positive cells in the intestines, significantly increases serum insulin, reduces blood glucose levels, and improves glucose tolerance[104].GLP-1 treatment induces insulin production in developing intestinal epithelial cells, and to a lesser extent, in adult intestinal epithelial cells bothin vitroandin vivo.The insulin+cells become responsive to a glucose challengein vitroand reverse insulin-dependent diabetes after implantation into diabetic mice[105].The GLP-1-induced conversion is mediated by the activation of Ngn3 and its downstream genes[104,105].We recently find that glucagon receptor mAb promotes intestinal L cell proliferation and increases circulating and intestinal GLP-1 levels in T2D mice.In addition, GLP-1 production is upregulated in the mouse L cell line and primary mouse and human enterocytes[92].These results suggest that glucagon receptor mAb might produce insulin+cells in the intestines.Other progenitors and mature cells, including hepatocytes and neuroendocrine cells, can also convert into β cells[83], but no clinical or preclinical drugs have been reported.
CONCLUSION
Pancreatic β cells own the ability of regeneration, and several types of cells can convert into β cells.The types of cells that can generate β cells, and the preclinical and clinical agents used for regeneration are summarized in Figure 1 and Table 1.Almost all hypoglycemic drugs and some immunomodulators can protect β cells by inhibiting β cell death and dedifferentiation through the correction of hyperglycemia and improvement of the consequent inflammation and oxidative stress.GLP-1 and other optimized molecules may promote stem cells to differentiate into β cellsin vitroand supplement β cell mass.Endogenous regeneration is an attractive approach.Regeneration in intraislets (including β cell proliferation and α/δ-to-β cell conversion), intrapancreas (including acinar-to-β cell reprogramming, reactivation of endocrine progenitor, and differentiation into β cells), and other sites (e.g., transdetermination of enteroendocrine progenitors to β cells) have all been investigated with some promising results.The different regeneration paths might represent different compensatory mechanisms for occasional β cell demise, relatively common stressors, or widespread islet loss and/or partial organ remodeling (as summarized in a previous review[69]).Notably, two or more regeneration paths often occur simultaneously in one condition.For instance, β cell replication and neogenesis from progenitors often occur together; α-to-β cell conversion generally induces progenitor activation.Notably, one drug often protects β cells from different aspects.For instance, GLP-1 and GABA promote the replication of β cells, enhance the conversion of α-to-β cells, and suppress immune reactions and cell apoptosis.As a result, GLP-1 and GABA have valuable significance for β cell regeneration and diabetes treatment.Several studies have identified the long-term effects of GLP-1.For example, exenatide improves β cell function for up to 3 years of treatment in patients with T2D[106,107].The safety and effects on the β cell function recovery of GABA in patients must be determined.
In addition, there are other areas that require further investigation.First, the regeneration mechanism between humans and rodents is different.Most strategies for promoting regeneration have only been successfully applied to animals and have failed in humans.Therefore, conclusions should be drawn cautiously from mouse models when interpreting the results for humans; moreover, carrying out experiments using human islets and conducting clinical trials would help.Second, the efficiency of β cell recovery induced by current clinical drugs is low, and other strategies that have high clinical efficiency and potential are required.Third, the win-win aim of hypoglycemia and islet regeneration is challenging when using a single agent, and a combination strategy is needed.A possible strategy might involve agents with good glucose-lowering efficacy and agents that have been demonstrated potential to preserve and regenerate β cells.On one hand, the agents with good glucose-lowering efficacy (such as insulin), achieve glycemic goals rapidly, thereby minimizing the exposure of β cells to glucotoxicity and lipotoxicity.On the other hand, several agents, such as GLP-1 or GABA, have been demonstrated to be potentially able to preserve and regenerate β cells, and may potentially contribute to the aim of recovering β cell mass.Fourth, the promotion of proliferation and neogenesis may lead to the development of cancer.The investigation of signals that mediate the physiological expansion of β cell mass in obesity and insulin resistance might lead to novel β cell regeneration reagents without significant tumorigenic risks.Besides, regulatory mechanisms to turn on and off regenerative and oncogenic pathways require investigations before applying regenerative approaches clinically.Additionally, further investigation is required into how the interventions to expand β cell mass can be specifically targeted to β cells.Taken together, in the last century, considerable efforts have been made to achieve complete β cell regeneration, and several agents have showed clinical potential, but there is still a long way to go.
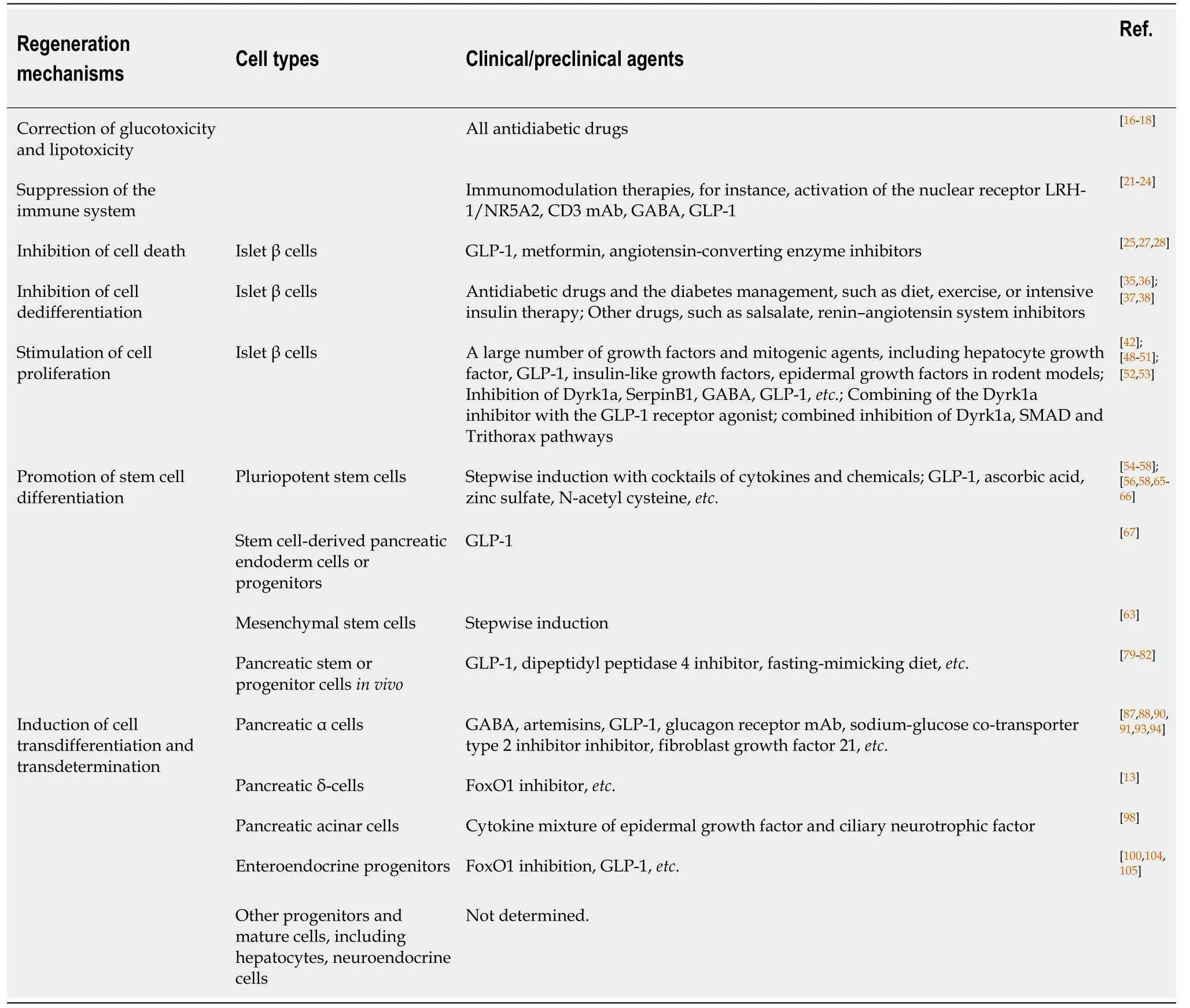
Table 1 Cell types, regeneration mechanisms, and clinical/preclinical agents for pancreatic β cell regeneration
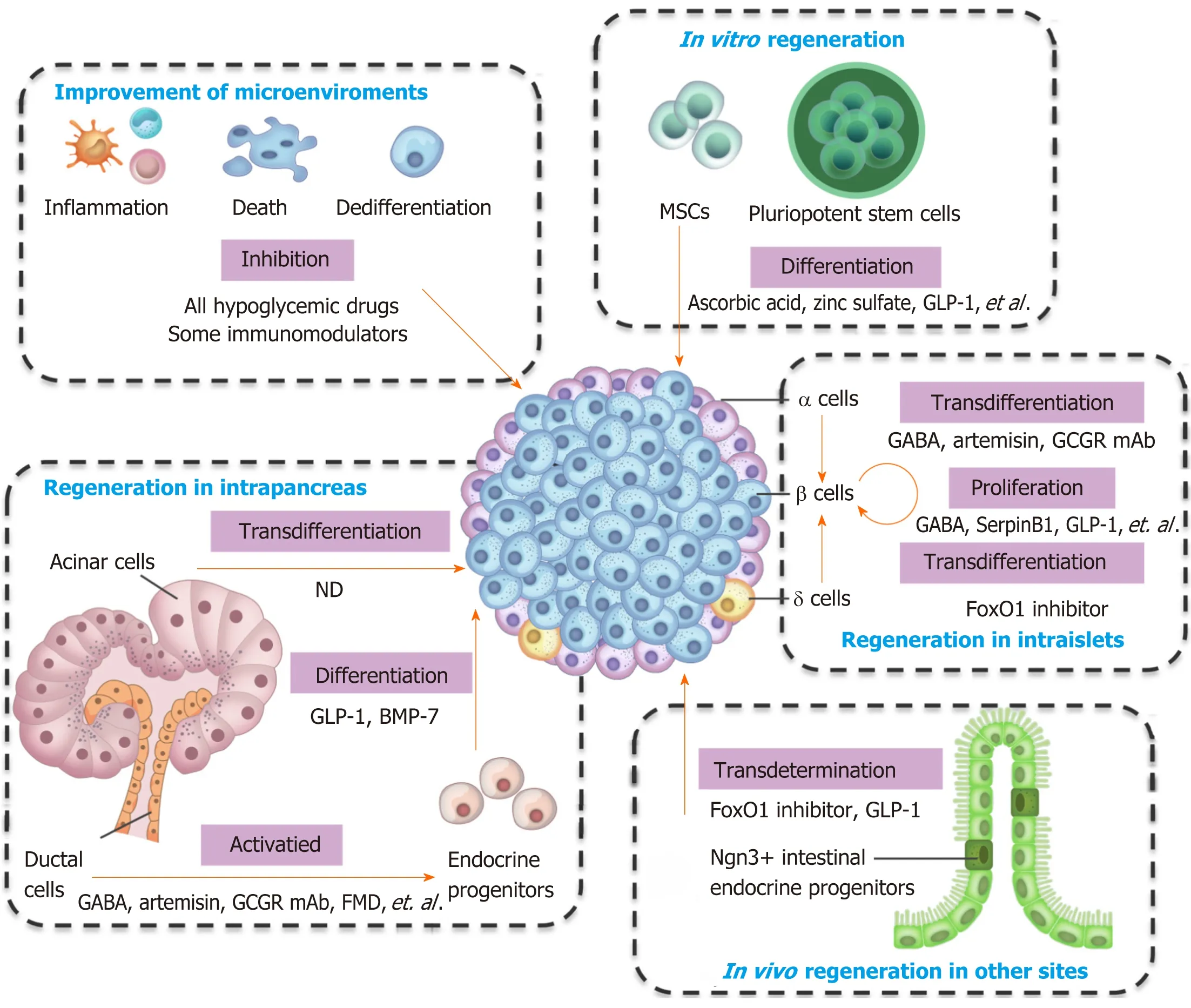
Figure 1 Integrative view of the cell types and clinical/preclinical agents for β cell regeneration.
杂志排行

World Journal of Stem Cells的其它文章
- Perspectives of pluripotent stem cells in livestock
- Adipose-derived stem cells: Pathophysiologic implications vs therapeutic potential in systemic sclerosis
- Mesenchymal stem cell-derived small extracellular vesicles in the treatment of human diseases: Progress and prospect
- Stem cell transplantation and/or adenoviral glial cell line-derived neurotrophic factor promote functional recovery in hemiparkinsonian rats
- Vascularization and osteogenesis in ectopically implanted bone tissue-engineered constructs with endothelial and osteogenic differentiated adipose-derived stem cells
- Proliferation and tenogenic differentiation of bone marrow mesenchymal stem cells in a porous collagen sponge scaffold