基于BSA重测序定位花生含油量相关基因位点
2021-02-07李玉颖于海洋吕玉英杨会张秀荣张昆刘风珍万勇善
李玉颖,于海洋,吕玉英,杨会,张秀荣,张昆,刘风珍,万勇善
(山东农业大学农学院/作物生物学国家重点实验室/山东省作物生物学重点实验室,山东 泰安 271018)
花生是全球范围内重要的油料作物,目前国产花生总量的55%用于榨油[1]。植物油脂是人类饮食的重要成分,含有的多种脂肪酸是维持人体生长、发育和机体正常活动的必需脂肪酸。此外,植物油脂中含有的不饱和脂肪酸可降低血清胆固醇和甘油三酯,能有效防治心脑血管疾病。植物油脂也是重要的能源和工业原料,是生物燃料的原料[2]。所以提高籽仁含油量是花生育种的重要目标之一。
植物种子油脂是由脂肪酸和甘油合成的高级脂肪酸甘油酯[3],以三酰甘油的形式储存于种子中,在成熟种子中主要以油体的形式存在。在脂肪酸合成途径中,乙酰CoA羧化酶(ACCase)催化的反应是脂肪酸生物合成的限速步骤,是控制碳流进入脂肪酸生物合成的重要调控位点[4]。脂肪酸合酶复合体中的酰基载体蛋白ACP也是脂肪酸合成中的重要蛋白[5,6]。脂肪酸和甘油一般通过Kennedy途径[7]形成三酰甘油,主要涉及3个酰基转移酶——磷酸甘油酰基转移酶(glycerol-3-phosph ateacyl transferase,GPAT)[8-10]、溶血磷脂酸酰基转移酶(lysophosphatidic acid acyltransferase,LPAAT)[11,12]、二酰甘油脂酰转移酶(diacyl glycerol acyl transferase,DGAT)[13,14]。
植物油脂合成代谢受到转录因子的调控,比如LEC、WRI、DOF[15,16]等。另外,种子发育阶段对油脂积累也有影响,种子发育早期油脂的积累比较缓慢,开花后21~39天是油脂含量增加最快的时期,种子成熟时油脂含量达到最大值[17]。唐兆秀等[18]发现花生荚果充实过程中,籽仁粗脂肪含量增长曲线呈抛物线型。同时,激素也可以调节油脂的积累。高浓度的ABA对于多数油脂合成途径中的基因表达具有抑制作用[19]。NAA诱导对脂质合成积累表现出显著的促进效应[20]。
综上所述,植物油脂合成代谢是重要的生物学过程,受到许多因素的影响,遗传机制复杂。利用QTL定位方法研究花生籽仁含油量并挖掘候选基因具有重要意义。近年来花生含油量相关QTL定位研究取得一定进展。郭建斌[21]在B03和A08染色体上检测到与含油量相关的2个QTLs,贡献率为9.76% ~22.00%。李新平等[22]在3个种植环境下检测到15个与含油量相关的QTLs,分布在LG1、LG7、LG8、LG15、LG16、LG18、LG19共7个连锁群上,贡献率为4.64% ~16.24%。Yaduru等[23]在A02、A08、A10、B03、B06、B09染色体上,共获得8个与含油量相关的QTLs,贡献率为5.67% ~22.11%。王瑶[24]在4个种植环境下共检测到10个含油量相关QTLs,分布在A02、A03、A04、A06、A07、B01、B02、B04染色体上,贡献率为2.36% ~11.61%。以上是通过构建遗传图谱进行连锁分析获得的QTL位点,这种方法成本高、耗时长。BSA(bulked segregant analysis)又叫集团分离分析法,是用于基因快速定位的一类分析方法,其选择群体中两极端性状个体构建混池,通过分析两极端混池的差异获得与性状相关联的分子标记。它不需要对所有个体进行分析,成本低、效率高,是目前基因定位的有效手段[25]。目前尚未见有利用BSA重测序对花生含油量相关基因定位的研究。本研究以本课题组选育的高油品系农大D666为父本,与普通含油量品种P12杂交,构建F2群体,利用BSA重测序方法分析定位与花生含油量相关的QTLs,为提高花生籽仁含油量及分子标记育种提供参考。
1 材料与方法
1.1 试验材料与性状调查
2017年以普通含油量品种P12为母本、高油花生品系农大D666为父本进行杂交,构建了包括568个单株的F2群体。按单株进行收获,荚果自然晒干。选取饱满均一的籽仁20 g磨成粉末,取2 g粉末为一个重复,共3次重复。采用索氏提取法[26]测定花生籽仁含油量。
1.2 极端混池构建与测序
根据索氏提取法测定的花生籽仁含油量数据,选取F2群体中极端高油和低油单株各30个,以及两个亲本P12和农大D666,取其幼嫩叶片后用DNA提取试剂盒提取基因组DNA。将30个高油单株和30个低油单株的DNA分别等量混合,构建高油和低油混池。对亲本及两个混池进行重测序,4个样本分别将DNA进行机械打断,对DNA片段进行修饰,再用琼脂糖凝胶电泳选择200~300 bp的片段,进行PCR扩增形成测序文库,文库质检合格后用Illumina HiSeq平台进行测序。对测序得到的原始数据进行过滤,主要步骤如下:(1)去除带接头(adapter)的reads;(2)过滤N含量超过10%的reads;(3)去除低质量reads(质量值低于10的碱基超过50%的reads)。另外,过滤掉同时比对到多条染色体上的reads。
利用bwa[27]软件将过滤后的reads比对到花生参考基因组(参考基因组下载网址:https://www.peanutbase.org/peanut_genome/)。比对结果使用Picard[28]的Mark Duplicate工具去除重复,使用GATK[29]软件进行SNP和InDel变异检测,对变异检测结果进行严格过滤,利用ANNOVAR[30]对SNP和InDel变异检测结果进行注释。
1.3 花生籽仁含油量性状的基因定位
筛选亲本间纯合差异的SNP位点,以高油亲本农大D666作为突变型,分析计算两个子代混池在亲本间标记位点的SNP-index(即SNP的频率)及 ΔSNP-index。计算方法如下:SNP-index(a/b)=Ma/b/(Ma/b+Pa/b),其中:Ma/b表示a/b池来源于突变性状亲本的深度,Pa/b表示a/b池来源于野生性状亲本的深度;ΔSNP-index=SNPindex(a)-SNPindex(b),其中:a代表突变性状对应的混池,b代表野生性状对应的混池。选择1 Mb为窗口、1 kb为步长对ΔSNP-index在各个染色体上的分布进行平滑作图,选取99%置信水平作为筛选的阈值,置信水平以上的窗口作为与含油量关联的区域。
同时使用ED算法分析与含油量关联的区域,利用两混池间差异的SNP位点,统计各个碱基在不同混池中的深度,计算每个位点的ED值,ED值越大表明该SNP在两混池间的差异越大。
ED值计算方法:
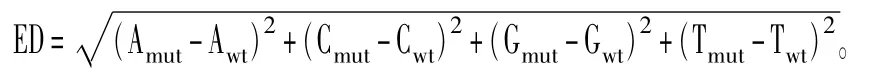
其中:Amut表示A碱基在突变性状混池中的深度,Awt表示A碱基在野生性状混池中的深度;Cmut表示C碱基在突变性状混池中的深度,Cwt表示C碱基在野生性状混池中的深度;Gmut表示G碱基在突变性状混池中的深度,Gwt表示G碱基在野生性状混池中的深度;Tmut表示T碱基在突变性状混池中的深度,Twt表示T碱基在野生性状混池中的深度。然而实际应用中,混池间的测序量差异会导致ED结果的偏倚,为了消除这种误差,本项目使用各位点上每种碱基的频率代替绝对深度值计算ED值,同时为消除背景噪音,对原始ED值进行5次方处理。然后采用局部线性回归LOESS方法,利用位点的位置信息进行拟合分析。拟合的方法为:每一个SNP拟合后的关联值等于前后各n个SNP的关联值的中值。取95%的置信水平作为分析的关联阈值。将超过阈值的区域筛选出来,可得到与含油量相关的区域。
将两种关联分析方法得到的区域进行比较,重叠区域作为与含油量相关的候选区域。利用基因组注释网站BLAST,对候选区间内的基因和多态性位点进行注释。
2 结果与分析
2.1 花生含油量性状统计分析
母本P12是粉红花皮小粒花生(图1),索氏提取法测定含油量为51.90%;父本农大D666是紫黑种皮小粒花生,索氏提取法测定含油量为55.84%,亲本之间含油量差异显著。利用索氏提取法测得F2群体单株含油量最大为61.91%,最小为47.04%,群体含油量变异范围较大,变异系数为3.90%(表1)。F2群体单株含油量性状表现为连续变异,符合正态分布,属于典型的数量性状(图2)。

图1 亲本籽仁比较
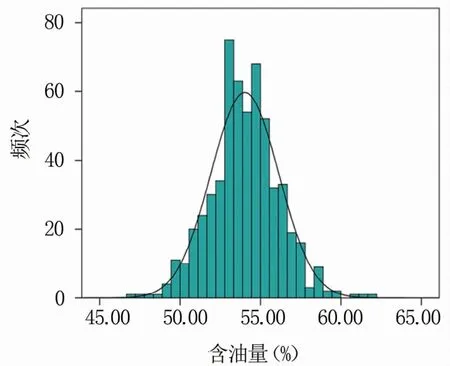
图2 F2群体含油量频次分布
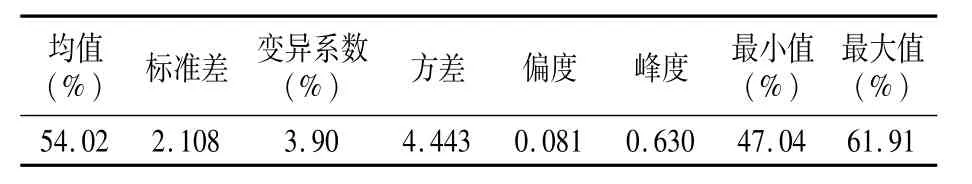
表1 F2群体含油量分布特性
2.2 全基因组重测序和SNP、InDel检测
根据测得的含油量数据,在F2群体中选取30个高油单株和30个低油单株分别构建高油和低油混池,并结合两个亲本进行BSA重测序分析。30个高油单株的含油量范围57.23%~61.44%,平均值为58.37%,30个低油单株的含油量范围47.04%~51.03%,平均值为49.83%。通过Illumina HiSeq平台对亲本及两个混池进行全基因组重测序,农大D666、P12、低油混池、高油混池测序数据量分别为81.87、82.90、89.69、90.25 Gb,过滤后得到的Clean Reads数 分 别 为342 714 021、341 342 887、375 501 075、378 106 976,P12和农大D666的平均覆盖深度为18×,两混池的平均覆盖深度为8×。4个样本测序数据Q30为88.74% ~90.24%,GC含量36.42% ~37.74%,与参考基因组的比对率均为100%(表2)。

表2 测序数据评估及与参考基因组比对统计
亲本P12和农大D666检测到的多态性位点分别为509 634、331 882个,检测到的SNP数目显著多于InDel。筛选亲本间纯合差异位点,共得到271 376个SNP和58 903个InDel。271 376个SNP中有1 885个位于外显子区,引起非同义突变的SNP共1 253个。58 903个InDel中有246个位于外显子区,引起移码突变的共239个。
2.3 与花生含油量关联的候选基因组区域
对于亲本间纯合差异SNP位点,以低油亲本P12为参考,计算两子代混池的SNP-index,则ΔSNP-index=SNPindex(高油混池)-SNPindex(低油混池)。ΔSNP-index越大,两混池差异越大,与花生含油量性状相关性越强。选择阈值以上的区域作为与花生含油量相关联的区域。在01号染色体上有3个超过阈值的区域,这些区域在基因组上的分布为Arahy.01:15173647~15536277 bp、Arahy.01:15641571~15989924 bp、Arahy.01:16007500~18384005 bp,把相距小于100 kb的区域合并,得到一个候选区域Arahy.01:15173647~18384005 bp(图3)。同时,进行ED关联分析,利用混池间差异SNP的深度差异,计算两混池间的ED值,结果显示分布于01、03、04、08、13、18共6条染色体的48个区域出现超过阈值的显著峰(图4)。

图3 两个子代混池ΔSNP-index在整个基因组上的分布

图4 两个子代混池ED值在整个基因组上的分布
把两种方法得到的重叠区域作为与花生含油量关联的候选基因组区域,即Arahy.01:15173647~18384005 bp,总长3.21 Mb,在该区域中共检测到318个SNP和53个基因。其中300个SNP分布在基因间区,9个SNP位于upstream,1个SNP位于5′UTR,6个SNP位于downstream,2个SNP位于内含子区。对候选基因组区域的编码基因进行多个数据库的深度功能注释,从基因表达产物参与的生物过程分析,有19个基因的表达产物与蛋白质的合成代谢有关,与氧化还原酶类相关的基因有4个,与跨膜转运相关的基因有3个,另外还有基因参与微管运动、细胞周期、糖类合成、信号转导等过程。
通过基因功能注释分析,在候选基因组区域内发现1个与脂质代谢相关的基因Arahy.55ECQ6。该基因位置是Arahy.01:15299758~15304403 bp,全长4 646 bp。Arahy.55ECQ6下游检测到1个SNP位点。该位点低油池SNP-index为0.200,高油池为0.818,ΔSNP-index值为0.618,说明两子代混池在该位点差异较大,可能与花生籽仁含油量有关。
3 讨论与结论
本研究利用普通含油量花生品种P12与高油花生品系农大D666杂交构建了1个F2群体,通过BSA重测序分析,获得1个与花生籽仁含油量相关的候选区域,在基因组上的分布为Arahy.01:15173647~18384005 bp,总长3.21 Mb,区间内检测到318个SNP和53个注释基因。李新平等[21]在A01染色体上也定位到了与花生含油量相关的QTL,但与本研究所得候选区域没有重叠,说明本研究获得的候选区域中存在新的QTL位点,可以进一步分析。
本研究预测的候选基因Arahy.55ECQ6编码磷脂酰肌醇特异性磷脂酶C(PI-PLC)X结构域结合蛋白。PI-PLC是一种真核细胞内酶。它催化1-磷脂酰-d-肌醇-4,5-二磷酸(PIP2)水解成甘油二酯(DAG)和肌醇-1,4,5-三磷酸(IP3)。甘油二酯(DAG)是合成三酰甘油(TAG)的底物。在二酰甘油脂酰转移酶(DGAT)的作用下,脂酰CoA加入到甘油的sn-3位上,生成三酰甘油。
本研究得到的候选基因组区域ΔSNP-index拟合曲线峰较平缓,可能由于该区域的基因位点是一个微效QTL。同时利用ED关联分析方法获得了多个超过阈值的显著峰,说明还存在其他含油量相关基因位点,为花生含油量相关基因定位研究奠定基础。
