解淀粉芽孢杆菌实时荧光定量PCR测定方法的建立及其在豆粕发酵中的应用
2021-02-06杨湘黔崔京春曾诗娴张珂彬胡晓红鲍雅静
杨湘黔 崔京春* 曾诗娴 成 丽 张珂彬 胡晓红 鲍雅静
(1.大连民族大学生命科学学院,大连116600;2.大连民族大学环境与资源学院,大连116600)
豆粕是大豆加工的副产物,因富含多种营养物质,是目前使用最广泛的植物性蛋白质饲料原料,但因豆粕中含有抗营养因子影响了动物对饲粮的消化吸收和利用[1-3]。解淀粉芽孢杆菌(Bacillusamyloliquefaciens)是一种广泛被用于开发微生物饲料添加剂的革兰氏阳性芽孢杆菌[4-5],它能够产生蛋白酶、纤维素酶、淀粉酶、脂肪酶等多种酶类,并能产生对动物病原菌中的大肠杆菌、梭状芽孢杆菌、沙门氏菌等具有较强的抑制能力的物质[6-8]。研究表明,利用解淀粉芽孢杆菌发酵豆粕不仅可以降低豆粕中的抗营养因子含量,且可提高动物对豆粕饲料的消化吸收率[9-12]。解淀粉芽孢杆菌在发酵豆粕饲料中的数量是影响豆粕发酵效果的关键因素之一,而由于用于发酵的豆粕为未灭菌原料,在进行接种菌的数量检测时会因自身带有的微生物的干扰,造成常用的微生物数量检测平板活菌计数法计数不准或者难以计数,并且由于该方法检测周期通常长达2~3 d,无法实现对豆粕发酵生产过程的即时监控[13-16]。
近几年,基于DNA水平上的微生物数量检测分子生物学方法越来越多,包括常规PCR技术、变性梯度凝胶电泳技术、实时荧光定量PCR技术等[17-22]。其中,实时荧光定量PCR技术具有特异性强、灵敏度高、操作时间短的特点,且由于在检测过程中均为闭管操作不会造成因污染而出现的假阳性结果[23-24]。DNA解旋酶A亚基(gyrA)基因是细菌DNA促旋酶上的1个亚基,该基因是枯草群中鉴定、分类的重要工具,因此,采用该基因可准确地区分枯草菌群中的近缘种[25]。
本研究拟建立基于解淀粉芽孢杆菌gyrA基因保守区设计引物、探针的实时荧光定量PCR方法,预期该方法可以达到种间特异,检出敏感度可满足监控解淀粉芽孢杆菌在不同豆粕发酵时期中的数量的要求,可以解决解淀粉芽孢杆菌发酵豆粕过程品质监控的瓶颈难题,并为解决以未灭菌生产原料发酵生产过程监控提供了有益的借鉴。
1 材料与方法
1.1 材料与试剂
1.1.1 试验菌种
本研究所选菌种为芽孢杆菌4株(解淀粉芽孢杆菌、地衣芽孢杆菌、蜡样芽孢杆菌、纳豆芽孢杆菌、枯草芽孢杆菌),非芽孢杆菌5株,其中包括乳酸杆菌2株(副干酪乳杆菌、植物乳杆菌),金黄色葡萄球菌、藤黄微球菌、大肠杆菌各1株。
1.1.2 试剂
dNTP、rTaq酶、10×Buffer、琼脂糖、6×Loading Buffer、2000 bp Marker、pMD18-T载体、TaKaRa Premix EX TaqTM购于宝生物工程(大连)有限公司;4S Green染料、IPTG即用型溶液、X-Gal即用型溶液、氨苄青霉素钠、DH5α感受态细胞、SanPrep柱式质粒DNA小量抽提试剂盒、Ezup柱式土壤DNA抽提试剂盒购于生工生物工程(上海)股份有限公司;TE缓冲液购于北京索莱宝科技有限公司。
1.2 试验方法
1.2.1 菌种培养
乳酸菌接种至MRS肉汤,其他菌株接种至牛肉膏蛋白胨液体培养基,37 ℃过夜培养。
1.2.2 菌种DNA的提取
纯菌液DNA的提取:以改进的水煮法提取纯菌液DNA[26],具体为:取过夜培养的菌液1 mL于1.5 mL离心管中,离心,弃上清并加入500 μL TE缓冲液,重悬菌体,99~100 ℃水浴15 min后离心,取上清作为DNA模板,-20 ℃保存,备用。
豆粕及发酵豆粕样品中DNA的提取:称取豆粕样品300 mg,采用试剂盒提取其DNA,具体操作步骤详见生工生物(上海)股份有限公司的Ezup柱式土壤DNA抽提试剂盒说明书。
1.2.3 特异性引物及探针的设计
在NCBI中的GenBank数据库中下载35株芽孢杆菌属的gyrA基因及16S rRNA基因全序列,利用DNAMAN软件对下载的序列进行比对,找出解淀粉芽孢杆菌的保守区,然后使用IDT引物设计软件进行引物、探针的设计,并对设计出的引物的退火温度、二聚体、发夹结构及探针的退火温度进行检测,从中筛选出1个正向引物、1个反向引物、1个探针(表1)。采用NCBI上的Primer-Blast Tool的引物比对软件从理论上确认引物、探针特异性,并由宝生物大连有限公司进行合成。该探针5’端由FAM基团(报告基团)修饰,3’端由TAMRA基团(淬灭基团)修饰。
1.2.4 常规PCR验证引物特异性及筛选
以1.2.2纯菌液中提取的10株菌的DNA为模板,以表1设计的引物进行常规PCR反应。以50 μL体系进行PCR扩增,其中包括10×Buffer(Mg2+free)5 μL,dNTP Mixture 4 μL,上、下游引物各1 μL,DNA模板2 μL,rTaq酶0.5 μL,无菌水补足至50 μL。PCR反应条件:94 ℃预变性5 min,94 ℃变性30 s,58 ℃退火30 s,35个循环;72 ℃延伸1 min,终延伸72 ℃ 5 min。PCR产物采用2%琼脂糖凝胶电泳进行检测。
1.2.5 实时荧光定量PCR方法的建立及特异性检测
实时荧光定量PCR反应体系:总体积为20 μL,其中包括premix EX TaqTM12.5 μL,上、下游引物(0.2 μmol/L)各0.5 μL,探针(0.2 μmol/L)0.8 μL,DNA模板2 μL,双蒸水补足至20 μL。
实时荧光定量PCR反应条件:预变性95 ℃ 30 s,变性95 ℃ 5 s,退火60 ℃ 34 s。特异性检测:以1.2.2纯菌液中所提取的10株菌的DNA作为模版,采用所建立的实时荧光定量PCR方法进行特异性验证。

表1 引物、探针序列
1.2.6 标准曲线的建立及灵敏性、重复性验证
pMD18T-gyrA基因重组质粒的构建:以解淀粉芽孢杆菌KCA基因组为模版及引物gyrA1-F/gyrA1-R进行扩增获得目的片段(115 bp),将其与pMD18-T载体连接,连接后的产物进行PCR和测序验证。验证正确后转化至大肠杆菌DH5α细胞中,然后采用SanPrep柱式质粒DNA小量抽提试剂盒提取重组质粒,通过核酸微量测定仪测定其浓度并通过公式换算成拷贝数。公式如下:
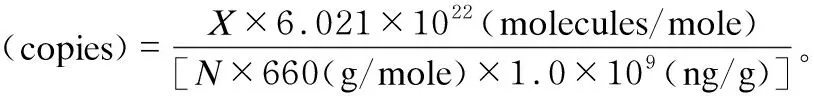
式中:X为重组质粒的所测浓度;N为重组质粒的碱基数。
标准曲线的建立:以洗脱缓冲液TE将标准品pMD18T-gyrA进行10倍稀释后作为模板进行实时荧光定量PCR反应。以拷贝数的对数为横坐标,Ct值为纵坐标建立标准曲线。扩增效率(E)计算公式为:
E=10-1/slope-1×100。
每个稀释度重复3次,以无菌水为阴性对照。
灵敏性验证:重组质粒标准品灵敏度检测是将标准品pMD18T-gyrA进行10倍梯度稀释并稀释成6个梯度,以作为实时荧光定量PCR的模版,不同稀释度的标准品重复3次检测;菌液灵敏度检测是将KCA菌原菌液进行10倍梯度稀释,分别提取各个稀释度菌液的基因组DNA,最后进行实时荧光定量PCR反应。
重复性验证:用5个稀释度的pMD18T-gyrA标准品的Ct值变异系数(CV)评价该方法的重复性。组内重复性验证分别以5个稀释度的标准品作为模板进行实时荧光定量PCR反应,每个稀释度重复3次,并以KCA菌的DNA和无菌水作为阳性对照和阴性对照。组间重复性验证以上述5个稀释度的标准品作为模板分3次进行实时荧光定量PCR反应,以KCA菌的DNA和无菌水作为阳性和阴性对照。
1.2.7 抗干扰验证
核酸水平抗干扰检测:分别提取108、103CFU/mL菌液量的解淀粉芽孢杆菌KCA基因组DNA,分别加入108CFU/mL菌液量的副干酪乳杆菌rg-1(Lactobacillusparacaseirg-1)所提取的DNA,以纯的解淀粉芽孢杆菌KCA的基因组DNA为对照进行实时荧光定量PCR检测,重复3次。
豆粕样品对接种菌吸附干扰的检测:取6 g灭菌的豆粕样品分别添加4 mL解淀粉芽孢杆菌KCA的不同稀释度菌液,静置10 min后加入20 mL磷酸盐缓冲液(PBS),颠倒2~3次后斜面振荡15 min,静置5 min后吸取上清,3 000×g离心6 min,弃上清,加入2 mL TE Buffer后提取基因组DNA;同时提取2 mL不同稀释度纯菌液的基因组DNA作为对照,并进行实时荧光定量PCR检测。每个稀释度检测3次。
1.2.8 发酵豆粕中解淀粉芽孢杆菌数量的检测
解淀粉芽孢杆菌KCA培养至108~109CFU/mL,以4%的接种量接种至水分含量为40%~45%的豆粕中,室温发酵0、1、2、3、5 d;取样根据1.2.2所提供的豆粕中DNA提取方法提取细菌DNA,利用所建立的实时荧光定量PCR方法进行检测。
1.3 数据统计分析
试验数据采用GraphPad Prism 7.00软件进行统计分析及作图。结果以平均值±标准差表示。
2 结果与分析
2.1 特异性验证
2.1.1 常规PCR法验证、特异性引物筛选
以gyrA及16S rRNA基因为靶标设计出的3对引物进行常规PCR扩增,由表2可知,只有引物对gyrA1-F/gyrA1-R扩增解淀粉芽孢杆菌KCA,采用gyrA2-F/gyrA2-R可同时扩增解淀粉芽孢杆菌KCA及地衣芽孢杆菌KCB;采用引物对16S rRNA-F/16S rRNA-R可将芽孢杆菌属内的菌株扩增出条带(164 bp)。这表明引物对gyrA1-F/gyrA1-R能特异性扩增解淀粉芽孢杆菌,因此选用gyrA1-F/gyrA1-R作为本研究建立实时荧光定量PCR方法的特异性引物对。
2.1.2 实时荧光定量PCR方法特异性的验证
利用引物对gyrA1-F/gyrA1-R及探针gyrA1进行实时荧光定量PCR反应,KCA菌在22~24个循环处出现扩增曲线,而其他4株芽孢杆菌、5株非芽孢杆菌及阴性对照则在35个循环后出现扩增曲线(图1)。这表明本研究所建立的方法对于KCA菌有较强的特异性。

表2 不同引物特异性检测
~分别为解淀粉芽孢杆菌KCA、4株芽孢杆菌、5株非芽孢杆菌属及阴性对照。
2.2 pMD18T-gyrA标准品的制备及标准曲线的建立
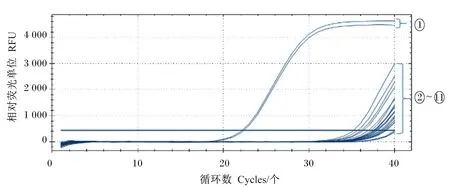
以KCA菌基因组DNA为模版,以引物对gyrA1-F/gyrA1-R进行常规PCR扩增,获得115 bp大小的片段(图2),结果与预期相符。将扩增产物与pMD18-T载体连接,构建重组质粒pMD18T-gyrA。构建后的质粒经酶切、测序验证正确后,使用核酸微量测定仪测定其浓度(结果未给出),并计算出其拷贝数为5.74×1010copies/μL,以此作为所建立的实时荧光定量PCR方法的标准品。
M为DNA Marker (2 000 bp);泳道1~3为gyrA1-F/gyrA1-R引物对的扩增产物;泳道4为阴性对照。
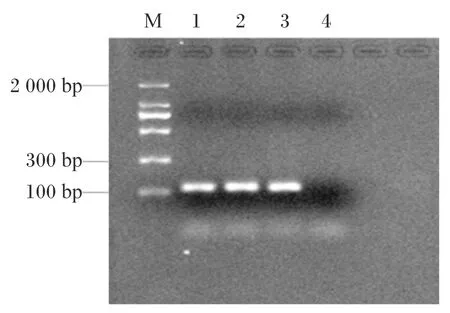
将标准品pMD18T-gyrA进行10倍梯度稀释,分别以各个稀释度的标准品作为模板进行实时荧光定量PCR反应,获得的扩增曲线规律较好,各稀释度之间的扩增Ct值相差在2~3个循环之间(图3);以不同稀释度的标准品拷贝数的对数为X轴、Ct值为Y轴建立标准曲线,该曲线的斜率为-3.468,扩增效率为0.94,截距为41.86,相关系数R2=0.999 3(图4),表明标准品浓度在5.74×107~5.74×102copies/μL有较好的线性关系。
2.3 实时荧光定量PCR方法的灵敏性及重复性检测
2.3.1 重组质粒标准品的灵敏度检测
以102~107copies/μL各个稀释度标准品pMD18T-gyrA为模板,利用所建立的方法进行检测。结果表明,该方法最小检出量为5.74×102copies/μL(表3)。
2.3.2 菌液灵敏度检测
将KCA菌的原菌液(4.0×108CFU/mL)进行10倍梯度稀释,从107~102各稀释度提取核酸作为模板,并进行实时荧光定量PCR反应。由表4可知,该方法的菌液最小检出限为103CFU/mL。
2.3.3 重复性检测
以不同稀释度的重组质粒标准品pMD18T-gyrA测定3次,组内重复Ct值的变异系数介于0.76%~3.27%,标准偏差(SD)介于0.10~0.55,组间重复Ct值的变异系数介于0.34%~1.88%,SD介于0.07~0.62,均在可接受范围内,证明建立的实时荧光定量PCR方法组内和组间重复较好。
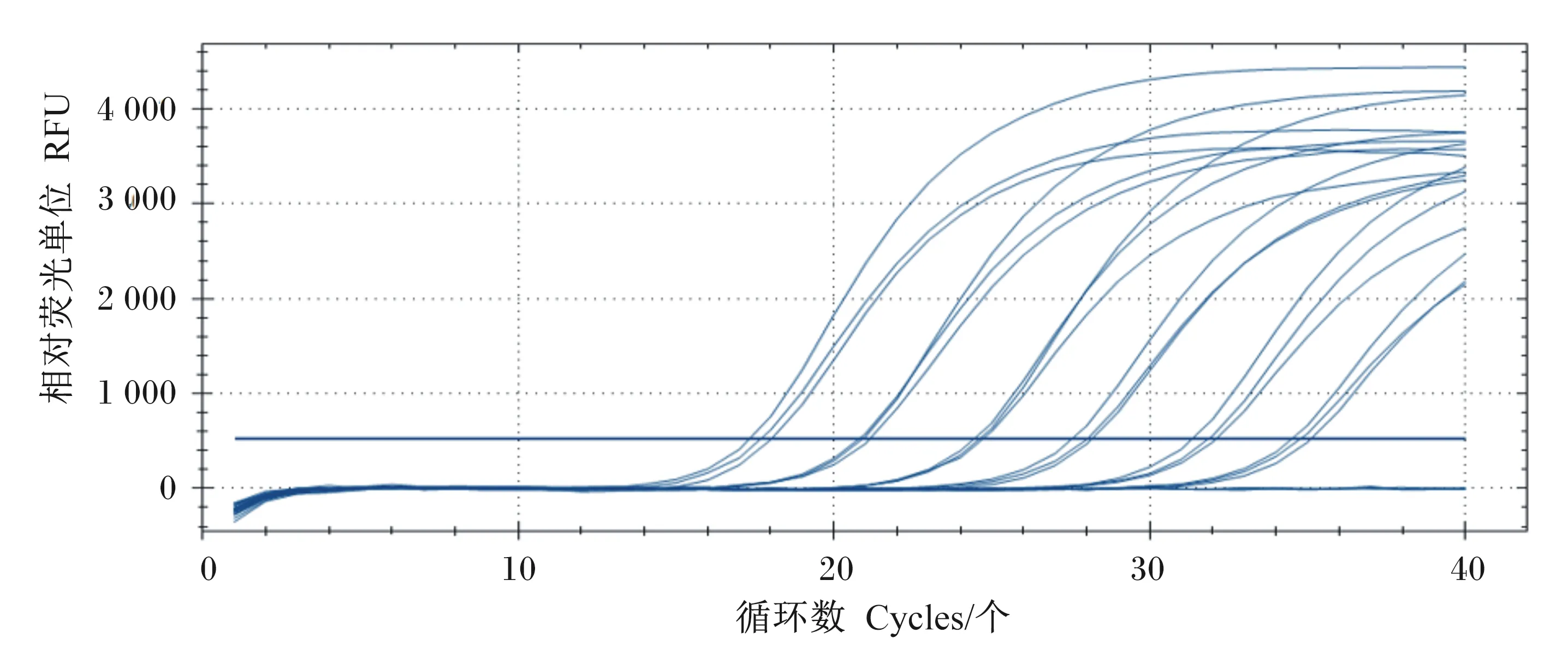
图3 不同稀释度标准品的扩增曲线

图4 实时荧光定量PCR标准曲线
2.4 实时荧光定量PCR方法的抗干扰验证
2.4.1 核酸水平抗干扰检测
由表5可知,从108和103CFU/mL菌液浓度
的KCA菌所提取的核酸中分别加入108CFU/mL的副干酪乳杆菌rg-1核酸提取物时(核酸浓度为6.7 ng/μL),KCA纯菌液的Ct值均不受影响,且与无rg-1干扰下的KCA纯菌液的Ct值无显著差异(P>0.05)。
2.4.2 豆粕对检测方法的干扰
由表6可知,纯菌液最低检测限为103CFU/mL,其中同等浓度下的纯菌液与豆粕的混合物的最低检出限为104~105CFU/mL,3次重复结果均比纯菌液检测限低1~2个梯度,表明以此方法检测发酵豆粕中目的菌的检测值比实际含量低10~100倍。

表3 实时荧光定量PCR标准品的灵敏度
2.5 发酵豆粕样品的实时荧光定量PCR检测结果
分别提取0、1、2、3、5 d的发酵豆粕样品基因组DNA作为模板,利用gyrA1-F/gyrA1-R引物对进行常规PCR检测。如图5所示,不同发酵天数的豆粕提取的DNA均有扩增条带,且随着发酵时间延长,条带越清晰,表明豆粕样品中的细菌DNA均提出。

表4 实时荧光定量PCR检测KCA菌液样品的灵敏度

表5 核酸水平抗干扰检测
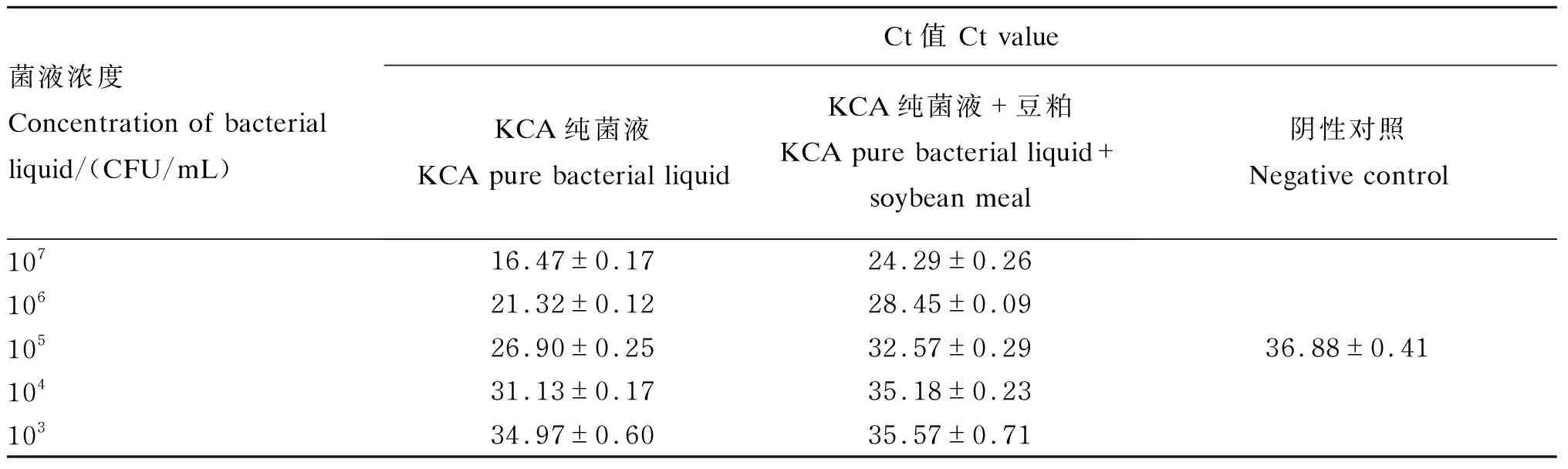
表6 豆粕对接种菌KCA检测的干扰
实时荧光定量PCR方法检测结果表明(表7),发酵0 d解淀粉芽孢杆菌数量为9.33×105copies/g(理论接种菌量为1.0×106CFU/mL),发酵5 d时,解淀粉芽孢杆菌的数量为4.16×108copies/g,此时的该菌的数量是发酵初期的1 000倍,说明本研究建立的实时荧光定量PCR方法能定量检测发酵豆粕中的解淀粉芽孢杆菌。
3 讨 论
解淀粉芽孢杆菌属于芽孢杆菌属(Bacillus),是枯草芽孢杆菌近缘种群的芽孢杆菌,该菌群中主要被用于微生物添加剂生产的近缘菌还有枯草芽孢杆菌(Bacillussubtilis)、地衣芽孢杆菌(Bacilluslicheniformis)[27-28]。细菌的16S rDNA序列素有“细菌化石”之称,已成为细菌系统发育分析的金标准。有研究表明,采用16S rDNA可以区分芽孢杆菌属内的菌种,但难以区分枯草近缘种群中的各类芽孢杆菌[29-30]。因此近几年采用细菌解旋酶gyrA基因进行细菌分类、鉴定颇受关注,Chun等[31]曾用gyrA基因设计枯草芽孢杆菌近缘种群的引物进行序列分析,表明gyrA基因能够区分枯草近缘种群中的芽孢杆菌。本研究针对解淀粉芽孢杆菌16S rRNA基因及gyrA基因的保守区设计了引物,常规PCR检测结果表明以gyrA基因设计出的引物进行扩增获得了良好的种间特异性,可将目的菌与近缘菌区别,而针对16S rRNA基因设计出的引物只能达到属间特异,这与曹凤明等[32]描述一致。
利用实时荧光定量PCR方法进行微生物的数量检测已在发酵领域得到较好的应用。Yong等[33]研究了固体发酵机制中产聚c-谷氨酸的解淀粉芽孢杆菌生长规律,并与活菌计数进行对比,表明建立的实时荧光定量PCR方法与活菌计数无显著差异;夏雪娟等[34]利用实时荧光定量PCR方法检测麻竹腌制过程中乳酸球菌的动态变化,结果表明随着腌制时间的延长,乳酸球菌数量逐渐升高,在14 d达到峰值(4.63×108copies/μL),相比于0 d(2.41×102copies/μL)增加了6个数量级。这些研究结果均表明,实时荧光定量PCR可以定量检测发酵过程中优势菌或接种菌的生长变化。本研究成功设计了可特异性检测解淀粉芽孢杆菌KCA的引物,并利用此引物扩增的产物构建的重组质粒,建立了标准曲线,建立了定量检测豆粕发酵过程中解淀粉芽孢杆菌KCA的实时荧光定量PCR的方法。该方法的标准品在5.74×107~5.74×102copies/μL有较好的线性关系,核酸水平最低检测限在102 copies/μL,纯菌液水平在103CFU/mL,说明该方法的灵敏性很好。此外,发酵豆粕的原料为未灭菌状态,从检样中提取细菌基因组DNA时通常为混菌的DNA,进行核酸水平、菌体水平抗干扰检测结果均表明所建立的方法抗干扰能力强,添加干扰菌核酸并不影响检测值。

M为DNA Marker (2 000 bp);泳道1为阴性对照;泳道2、3、4、5、6分别为发酵0、1、2、3、5 d豆粕样品。

表7 不同发酵时间的豆粕中解淀粉芽孢杆菌的数量检测
在实际发酵生产中,豆粕为干燥的粉末状,加菌发酵时会对目标菌造成吸附,从而导致在提取细菌DNA时会有残留,为了探讨豆粕类固体基质对样品中DNA检测的影响,本研究还进行了豆粕的抗干扰验证试验,结果表明豆粕中目的菌的定量检测限为104~105CFU/mL,比纯菌液的高10~100倍,说明豆粕类固体基质对接种菌的DNA水平数量检测有干扰,但因发酵豆粕时是以108~109CFU/mL的菌液、4%的接种量进行的,接种之初豆粕样品中目的菌的含量至少在106CFU/mL,检测基线比所建立方法高出1~2个数量级以上,不影响该方法在实际检测中的应用。实际检测结果表明,不同发酵时期的豆粕中的解淀粉芽孢杆菌数量均发生变化,发酵初期(0 d),解淀粉芽孢杆菌的数量为9.33×105copies/g,发酵至5 d,解淀粉芽孢杆菌的数量为4.16×108copies/g,是接种初期的1 000倍。同时表明解淀粉芽孢杆菌的数量随着发酵天数的延长而增加,进而降低豆粕中的抗营养因子含量,提高营养物质消化利用率,这与王梅等[35]描述一致。以上结果证明本研究建立的解淀粉芽孢杆菌的实时荧光定量PCR的方法可良好地应用于豆粕发酵过程中解淀粉芽孢杆菌的定量检测。
4 结 论
本研究针对发酵豆粕中接种菌建立了特异性实时荧光定量PCR方法,可相对准确检测接种菌在发酵豆粕中不同时期的数量变化,解决了以豆粕等未灭菌生产原料进行固体发酵过程中监控接种菌生长情况难的瓶颈问题,为发酵豆粕生产企业进行生产过程品质监控提供了新的科学的技术手段。
