In 掺杂h-LuFeO3 光吸收及极化性能的第一性原理计算*
2021-02-06张小娅宋佳讯王鑫豪王金斌钟向丽
张小娅 宋佳讯 王鑫豪 王金斌 钟向丽
(湘潭大学材料科学与工程学院,湘潭 411105)
h-LuFeO3 是一种窄带隙铁电半导体材料,已被证明在铁电光伏领域有较好的应用前景.然而,较低的极化强度使光生电子-空穴对复合率大,限制了h-LuFeO3 基铁电光伏电池效率的提高.为改善h-LuFeO3 的极化强度,提高光吸收性质,本文利用第一性原理计算方法研究了In 原子在h-LuFeO3 不同位置的掺杂形成能,得到最稳定的掺杂位置,比较了h-Lu1–xInxFeO3 (x = 0,0.167,0.333,0.667)的带隙、光吸收性能及极化强度等性质.计算结果表明,随着In 掺杂比例的增加,h-LuFeO3 的晶格常数c/a 比不断增大,铁电极化强度得到提高.当In∶Lu = 2∶1 时,材料杂质能级出现,Fe-O 轨道杂化得到增强,提高了h-LuFeO3 的光吸收性能.此工作证明了In 掺杂是改善h-LuFeO3 极化强度和光吸收系数的有效方法,对铁电光伏性能的提高提供一种新途径.
1 引 言
铁电材料中的铁电极化可作为内建电场促进光生载流子的分离,使铁电光伏电池能够实现光学带隙以上的开路电压[1−4].这种特殊的铁电光伏效应,为突破传统p-n 结型光伏电池的理论转换效率瓶颈提供了全新的方法,在新能源领域表现出巨大的应用潜力[5].研究人员对众多铁电材料,例如钛酸钡(BaTiO3)、锆钛酸铅(Pb(ZrxTi1–x)O3)、铌酸锂(LiNiO3)等的光伏性质进行了大量研究[6−8],然而,较大的带隙使得大部分太阳光能量无法被吸收(太阳光能量范围约为0.4—4 eV)[9].h-LuFeO3是一种新型多铁材料,光学带隙在2.0 eV 左右[10],在铁电光伏领域有较好的应用前景.由于本征h-LuFeO3较低的极化强度(仅为4.7 µC·cm–2)[11],使得h-LuFeO3基铁电光伏电池的光生载流子复合率较大,不利于电池光电转换效率的提高.目前实验上制备的h-LuFeO3基铁电光伏电池的光电转化效率在0.001%左右[12].先前的研究表明,提高h-LuFeO3的极化强度能够有效降低光生载流子的复合率,增加电池的开路电压[5],改善其光吸收性质,提高h-LuFeO3基铁电光伏电池的性能.
室温条件下h-LuFeO3的铁电性不稳定,可以通过应力使其稳定存在[13,14].然而采用外延应力调控h-LuFeO3铁电性能对实验条件的要求十分苛刻[15],极大地限制了应力对h-LuFeO3的调控效果.研究发现,采用掺杂的方法不仅能对h-LuFeO3性能进行调控,且能制备出铁电性在室温下稳定存在的h-LuFeO3[16−18].该方法对实验条件依赖小,能够增强材料的铁电光伏相关性能.在Lu 位置置换离子半径较小的离子,是获得稳定六角结构LuFeO3的一种重要方法.Lin 等[16]通过第一性原理计算表明,Sc 掺杂可以使LuFeO3更稳定,而其多铁性不受影响.但In 离子半径比Lu 和Sc 都要小,理论上InFeO3也是六角结构.Liu 等[17]利用In掺杂h-LuFeO3能得到六角结构的h-Lu1–xInxFeO3材料,X 射线全谱拟合结果表明,随着In 含量的增加,材料结构逐渐由极性的P63cm(x= 0.4—0.6)转变为非极性的P63/mmc(x= 0.75),即顺电相.而目前并未深入研究In 掺杂对h-LuFeO3光学性能的影响.本文基于密度泛函理论(density functional theory,DFT)的第一性原理计算方法对h-Lu1–xInxFeO3(x= 0,0.167,0.333,0.667)的磁结构、带隙、光吸收系数及自发极化等性质进行了研究,揭示In 掺杂对h-LuFeO3的极化性质及光学性质的影响,为提高h-LuFeO3的铁电光伏性能提供可靠的理论依据.
2 计算方法
使用的第一性原理计算方法均基于Materials Studio 软件的CASTEP 模块[10],采用广义梯度近似(generalized gradient approximation,GGA)和平面赝势波的方法处理电子与离子间的相互作用,利用GGA 下的Perdew-Burke-Ernzerhof (PBE)泛函作为交换关联泛函[19].赝势选取Ultrasolft,截断能为572 eV,计算中所有晶胞和原子都进行弛豫,以达到最稳定状态.电子自洽精度为10–5eV/atom,原子受力小于0.05 eV/Å,晶胞结构优化时第一布里渊区的k点采用4×4×2,电子结构优化时k点采用6×6×3.对于磁性元素Fe 采用GGA+U的方法来处理,其中U是位库仑势.参考先前的报道,U值选择为4.5[20].
3 结果与讨论
3.1 晶体结构
In 在h-LuFeO3的掺杂位置有两种,分别记为P1 和P2[21],如图1(a)所示.在包含30 个原子的晶胞模型中,有4 个P1 位原子和2 个P2 位原子,首先讨论h-Lu1–xFexO3最稳定的磁序.采用A 型、C 型、G 型三种反铁磁结构和铁磁结构对相同的晶胞模型进行优化.为了得到最稳定的晶胞结构,以G 型反铁磁序h-Lu0.833In0.167FeO3能量为基准,比较了不同掺杂位置的In 原子替换单个Lu 原子对h-LuFeO3磁序的影响,并比较了相对总能量,结果如表1 所列.结果表明,四种磁序中非线性G 型反铁磁序的h-Lu0.833In0.167FeO3能量最低,这与先前报道的计算结论一致[22],且无论是P1 位掺杂还是P2 位掺杂均没有改变磁序.利用(1)式计算了不同掺杂位置的掺杂形成能:

其中Ef为In 掺杂形成能,Edoped和Epristine分别代表掺杂后和掺杂前的h-LuFeO3总能,µLu和µIn分别为Lu 和In 的化学势.形成能反映了掺杂的难易程度,形成能越小越容易掺杂.通过计算发现,P1位置的掺杂形成能为5.426 eV,P2 位置的掺杂形成能为5.581 eV,表明P1 位置相对P2 位置更容易掺杂.

图1 h-Lu1–xInxFeO3 的球棍结构模型图 (a) P1,P2 位置;(b) h-Lu0.667In0.333FeO3; (c) h-Lu0.333In0.667FeO3Fig.1.Model of the ball-and-stick structure of h-Lu1–xInxFeO3: (a) P1,P2 position; (b) h-Lu0.667In0.333FeO3;(c) h-Lu0.333In0.667FeO3.

表1 不同磁序下不同位置的In 掺杂相对能量变化Table 1.Relative energy changes of In doping at different positions under different magnetic orders.
由于本征InFeO3稳定相是顺电相P63/mmc,计 算 时 控 制 了In∶Lu 最 高 为2∶1.h-LuFeO3的P1 位置比P2 位置更容易掺杂,故均选择In 原子替换P1 位原子,In∶Lu 分别为1∶2 和2∶1 时,对应的晶胞优化结果如图1(b)和图1(c)所示.以非线性G 型反铁磁序的h-Lu0.833In0.167FeO3能量为基准,比较了不同磁序的晶胞结构优化后相对能量的变化,结果如表2 所列.随着掺杂浓度的提高,h-Lu1–xInxFeO3(x= 0.333,0.667)磁序未发生变化,非线性G 型反铁磁序的h-Lu0.833In0.167FeO3能量最低.

表2 In∶Lu 为1∶2 和2∶1 时h-Lu1–xInxO3 不同磁序的相对能量Table 2.Relative energy of h-Lu1–xInxO3 with different magnetic sequencewhen In∶Lu is 1∶2 and 2∶1.
确定了最稳定的磁序后,计算P1 位的In 掺杂对h-LuFeO3晶格参数的影响,与先前实验报道的h-LuFeO3晶格参数进行对比,结果如表3 所列,表明非线性G 型反铁磁序h-Lu0.833In0.167FeO3结构优化结果与实验值符合较好.随着In 原子含量的提高,h-Lu1–xInxFeO3晶胞沿c轴拉伸,c/a比从1.94 增加到2.04,这有利于材料自发极化率的增强.而晶胞略微缩小,我们猜测可能是由于In 的离子半径比Lu 离子半径小造成的.
3.2 电子结构
h-Lu0.833In0.167FeO3,h-Lu0.667In0.333FeO3以及h-Lu0.333In0.667FeO3的能带图见图2.从图2(a)可知,h-Lu0.833In0.167FeO3的带隙大小为1.16 eV,小于实验值(2.0 eV)[24],与先前文献中利用了Local Density Approximation (LDA),Perdew-Burke-Ernzerhof for solid (PBEsol)等方法计算的h-Lu FeO3带隙对比如表4 所列.带隙偏小是因为利用DFT 方法会低估材料带隙,然而掺杂计算的主要目的是为了预测相对变化趋势,而非研究绝对值的大小,因此选取DFT 方法并不影响最终结论.
三种计算模型的h-Lu1–xInxFeO3导带底和价带顶均在G点,表明h-Lu1–xInxFeO3为直接带隙半导体材料.如图2(b)所示,当In∶Lu = 1∶2 时,即In 占据晶胞中两个P1 位置时,带隙减小到1.05 eV,表明In 原子的替换可减小材料的带隙.由图2(c)可知,当In∶Lu = 2∶1 时,In 完全占据4 个P1 位置,带隙的变化并不明显,仅下降了0.01 eV;但此时导带底(1—2 eV 范围内)杂质能级的增多有利于提升材料的光吸收系数.
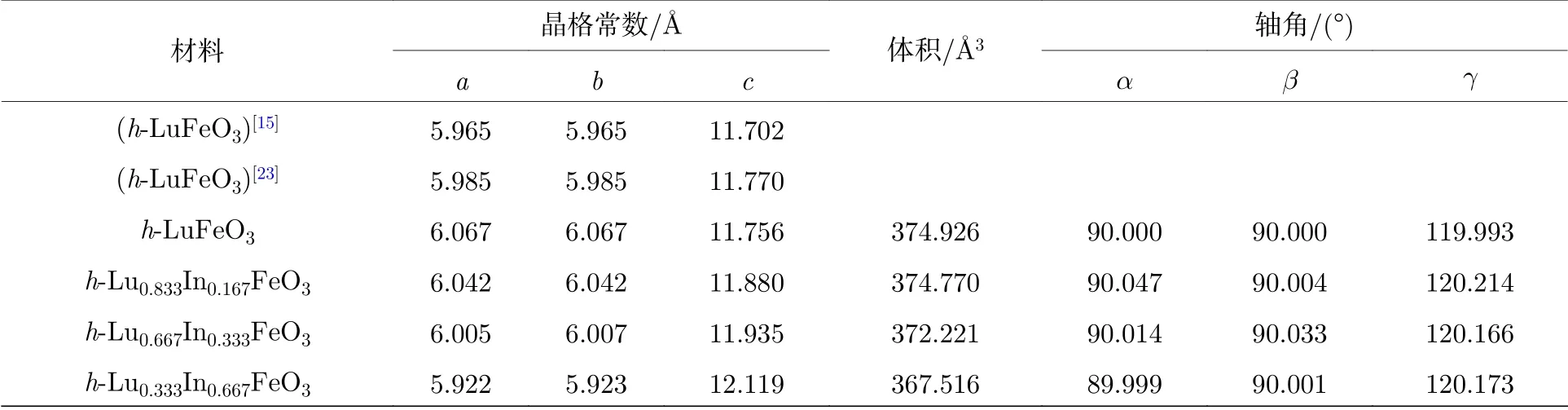
表3 h-Lu1–xInxO3 的结构优化结果Table 3.Structure optimization results of h-Lu1–xInxO3.

图2 h-Lu1–xFexO3 的能带图 (a) h-Lu0.833In0.167FeO3; (b) h-Lu0.667In0.333FeO3; (c) h-Lu0.333In0.667FeO3Fig.2.Energy band diagrams of h-Lu1–xFexO3: (a) h-Lu0.833In0.167FeO3; (b) h-Lu0.667In0.333FeO3; (c) h-Lu0.333In0.667FeO3.

表4 本工作带隙计算结果与已发表结果对比Table 4.Comparison of calculated band gap results with published results.

图3 分布态密度图 (a) 未掺杂的h-LuFeO3; (b) h-Lu0.333 In0.667FeO3Fig.3.Distribution density of states: (a) Undoped h-LuFeO3;(b) h-Lu0.333In0.667FeO3.
为了深入研究In 掺杂对h-LuFeO3带隙两端电子跃迁的影响,分析了未掺杂h-LuFeO3和h-Lu0.333In0.667FeO3在带隙附近,即能量在–2—2 eV 间的态密度图,如图3 所示.由图3(a)可知,未掺杂h-LuFeO3的导带底(1—2 eV 范围内)大部分由Fe 3d 轨道占据,小部分由O 2p 轨道占据;价带顶(–1—0 eV 范围内)主要是O 2p 轨道占据,而Lu 对价带顶和导带底的贡献有限,从主要占据价带顶的O 2p 轨道电子转变为主要占据导带底的Fe 3d 轨道电子,表现出Fe 3d 轨道与O 2p 轨道的高度杂化.因此调控Fe 3d 轨道与O 2p 轨道杂化程度可以影响材料的光吸收性能.
图3(b)为h-Lu0.333In0.667FeO3的态密度图,导带底(1—2 eV 范围内)大部分由Fe 3d 轨道占据,小部分由O 2p 轨道及In 4s 轨道占据; 价带顶(–1—0 eV 范围内)主要由O 2p 轨道占据.相对未掺杂的h-LuFeO3,h-Lu0.333In0.667FeO3杂质能级的出现及导带底Fe 3d 轨道赝势峰强度的增大,使材料带隙缩小,接收价带电子的能力增强.分析结果表明,In 掺杂影响了材料Fe-O 间的轨道杂化,提高了h-Lu0.333In0.667FeO3的光吸收系数.

图4 In 掺杂前后h-LuFeO3 光学吸收系数随入射光子能量的变化Fig.4.Change of optical absorption coefficient of h-LuFeO3 with incident photon energy before and after In doping.
3.3 光吸收性能及铁电极化强度
图4为光沿c轴入射h-Lu1–xInxFeO3的光吸收系数变化情况,发现在1.64—2.4 eV 范围内,掺杂后的光吸收系数比未掺杂的h-LuFeO3略微减小.但在能量大于2.4 eV 区域,材料光吸收系数随着In/Lu 比值的增大而增大,表明In 掺杂能够有效提高h-LuFeO3在太阳光能量范围内的光吸收效率.
铁电极化是影响铁电光伏性能的另一个重要性质.铁电光伏材料以自身铁电极化为内建电场,增大铁电极化能够减少载流子复合概率[26],因此缩小带隙和增大铁电极化强度是提高铁电光伏材料性能的两大研究方向.根据伯恩有效电荷定性计算自发极化强度计算公式为[27]

其中e表示电子电荷量,表示伯恩有效电荷,Δujβ表示原子相对位移,Ω表示晶胞总体积.将h-Lu1–xInxFeO3沿c轴的伯恩有效电荷代入(2)式,计算得到h-LuFeO3,h-Lu0.833In0.167FeO3,h-Lu0.667In0.333FeO3及h-Lu0.333In0.667FeO3沿c轴的铁电极化强度,分别为3.93,5.91,7.92 和11.02 µC·cm–2,如图5 中红色曲线所示.图5 中蓝色曲线表示随In/Lu比的增大,h-Lu1–xInxFeO3晶胞的晶格参数c/a比值的变化.可见材料的极化值与c/a比有相同的变化趋势,结果表明在保持铁电相的前提下,提高材料的c/a比可提高材料的极化强度.对铁电光伏材料而言,极化强度的增大能够得到更高的光生电压及光生电流[11].此外,改变c/a比相当于材料受力沿c轴产生拉应变,而拉应变能够减小六角铁酸盐类多铁材料的带隙并增强光吸收性质[13].

图5 不同In/Lu 比的h-Lu1–xInxFeO3极化值(红色曲线)和晶格常数c/a 比(蓝色曲线)Fig.5.Polarizationvalues(redcurve)andlatticeconstant c/a ratios (blue curve)of h-Lu1–xInxFeO3 with different In/Lu ratios.
4 结 论
本文基于DFT的第一性原理计算方法,研究了h-Lu1–xInxFeO3(x=0,0.167,0.333,0.667)的带隙、极化强度及光吸收性质.研究结果表明,In原子在h-LuFeO3晶胞中会优先替换P1位置,随着In 掺杂量的增加,h-Lu1–xInxFeO3的晶胞沿c轴拉伸.当x达到0.667 时,晶格常数c/a比从1.94增大至2.04.通过分析h-LuFeO3及h-Lu0.333In0.667FeO3的分态密度可知,c/a比的增大能够增强h-Lu0.333In0.667FeO3层间的Fe-O轨道杂化程度,提高在太阳光范围内的光吸收系数.利用伯恩有效电荷定性地计算h-In1–xLuxFeO3的极化强度,发现In掺杂能够提高材料沿c轴的极化强度,且极化值的变化趋势与晶格常数c/a比的变化趋势相同.因此,In 掺杂能够有效提高h-LuFeO3的光吸收系数及铁电极化强度,为提高铁电光伏性能提供重要理论指导.
