加权基因共表达网络分析鉴定阻塞性睡眠呼吸暂停的关键基因
2021-02-05曹媛媛蔡昕添汪思敏努尔古丽买买提李南方
曹媛媛 蔡昕添 汪思敏 洪 静 努尔古丽·买买提 李南方
阻塞性睡眠呼吸暂停(obstructive sleep apnea, OSA)是一种以夜间睡眠打鼾、呼吸暂停和日间嗜睡为特征的睡眠呼吸紊乱疾病,可导致间歇性低氧血症、高碳酸血症和睡眠结构紊乱[1]。与OSA相关的主要临床风险是多器官系统损害,如心脑血管疾病、代谢综合征和认知功能障碍等[1~3]。OSA在普通人群中的发生率呈逐年上升趋势,据估计,中度至重度OSA在男性中高达49.7%,在女性中高达23.4%[4]。然而,绝大多数OSA患者(70%~90%)没有得到及时诊治,造成了沉重的健康和社会经济负担[5]。因此,研究OSA的病因和发病机制,寻找早期诊断指标和治疗靶点至关重要。
据报道,OSA有较强的遗传影响,患者一级亲属的风险增加了1.5倍以上[6]。衡量OSA严重程度的呼吸暂停低通气指数(AHI)中约35%~40%的变异可以用遗传因素来解释[7]。目前,基于高通量测序的全基因组DNA微阵列已经成为研究复杂疾病遗传学的一种有效且相对经济的工具[8]。虽然已经发现了一些OSA分子标志物,但由于OSA的异质性及其复杂的病理生理状况,单个基因并不能准确地代表OSA的特征[7,9]。与侧重于单个基因的差异表达分析不同,共表达网络分析通过以无监督的方式识别协同表达的基因模块,为理解疾病的发病机制和治疗干预机会提供了新的见解[10,11]。它已被成功地应用于严重哮喘、慢性阻塞性肺疾病(COPD)及癌症等多种生物学过程的研究,在识别候选生物学标志物和治疗靶点方面被证明是相当有效的[11,12]。
本研究应用加权基因共表达网络分析(WGCNA),识别与OSA相关的共表达模块,对其生物学功能和通路进行注释;鉴定关键模块中的枢纽基因并深入分析,为寻找与OSA发病相关的潜在靶基因提供理论依据。
材料与方法
1.数据的获取:从NCBI的GEO数据库(http:∥www.ncbi.nlm.nih.gov/geo/)中下载OSA的全基因组表达数据集GSE135917进行初步分析,该数据集基于GPL6244平台,来源于10例OSA患者和8例正常对照者的皮下脂肪组织样本,其中包含了每例样本对应的年龄、性别、体重指数(BMI)及疾病状况的临床信息[13]。另一基于GPL6244平台的独立数据集GSE38792,包含10例OSA患者和8例正常对照者的内脏脂肪组织样本,用于后续验证[14]。
2.数据的处理与筛选:应用R语言及其程序包对含有原始数据(.CEL文件)进行预处理、归一化和质量控制,采用RMA法进行背景校正、分位数归一化和中值抛光。根据注释平台,将探针ID转换为基因符号。利用Limma包,以经典贝叶斯t检验进行OSA与正常组之间的差异表达分析,以Benjamini-Hochberg法校正P值为错误发现率(FDR)。本研究以|log2Fold Change|≥0.585且FDR<0.05为标准筛选出差异表达基因(DEGs)用于共表达网络的构建。
3.加权基因共表达网络的构建:通过R语言中的WGCNA包构建DEGs的共表达网络[11]。利用hclust函数进行聚类分析,剔除数据集中的离群样本,利用pickSoftThreshold函数确定合适的软阈值(β),得到拟合指数R2>0.8的近似无尺度网络分布。利用blockwiseModules函数进行一步法网络构建和模块检测,生成最小模块大小为30,合并切割高度为0.25的共表达基因模块与拓扑重叠矩阵(TOM)。对每个模块进行主成分分析,以第一主成分计算基因模块的特征值(module eigengenes, MEs)。引入上述临床表型,计算MEs与各临床表型之间的相关系数,识别与OSA显著相关的基因模块。模块内分析基因表达与表型的相关性(gene significance, GS)、与模块的相关性(module membership, MM),以筛选关键枢纽基因。
4.关键模块的功能富集分析:选取与OSA显著相关的基因模块,利用Cytoscape 3.7.2软件中ClueGo插件进行基因本体论(GO)注释、京都基因与基因组百科全书(KEGG)通路分析(kappa score=0.4,P≤0.01)。利用STRING 11.0数据库进行模块内基因的蛋白-蛋白相互作用(PPI)分析(combined score≥0.7),利用Cytoscape软件中cytoHubba插件的Degree法可视化排名前30位的基因(Top30)。
5.关键基因的识别与验证:从关键模块Top30基因构建的PPI网络中,依据节点度值选取排名前3位的基因为关键基因。整合GSE135917和GSE38792数据集,利用ggpubr、ggplot2包对关键基因在OSA和正常组织的表达再次验证,利用pROC包绘制ROC曲线评估关键基因的诊断价值和预测OSA的最佳截断值。
结 果
1.芯片数据的基本信息:经过数据预处理,从18个组织样本中共获得23281个基因表达值。以|log2Fold Change|≥0.585且FDR<0.05为标准,OSA与正常组相比,共筛选到3425个DEGs,其中上调基因497个,下调基因2928个。火山图显示了DEGs的分布,热图显示了DEGs与样本的双向分层聚类结果。为降低噪音及计算机的运行负荷,将3425个DEGs用于下一步共表达网络的构建(图1)。
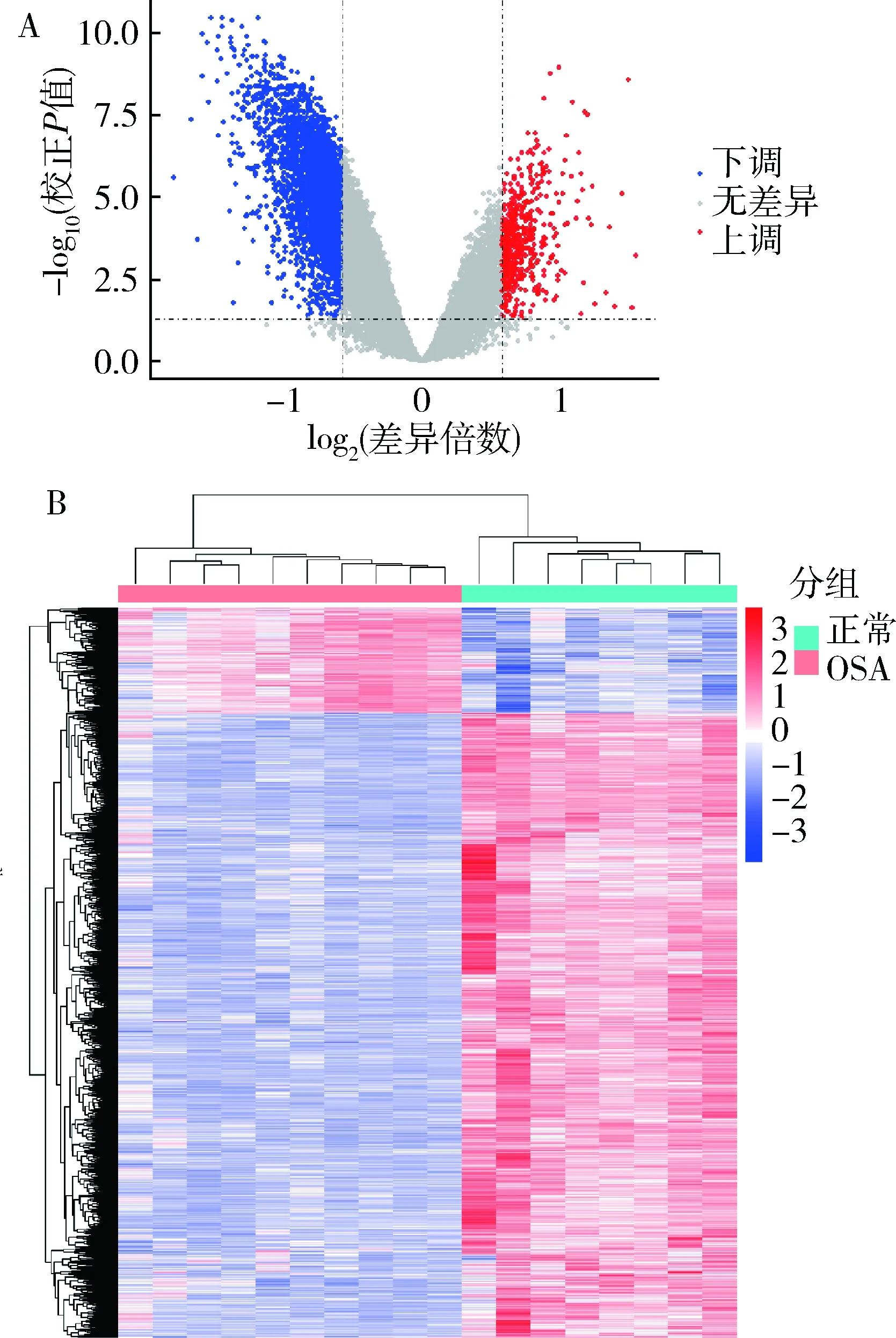
图1 GSE135917差异基因的筛选 A. 火山图(红点表示上调的差异基因;蓝点表示下调的差异基因;灰点表示无差异的基因)B. 热图(红色表示差异基因的高表 达;蓝色表示差异基因的低表达)
2.加权基因共表达网络的构建:通过WGCNA算法,依据无尺度网络分布拟合,选取 β=18作为本数据集的软阈值,并计算基因间的邻接矩阵与拓扑重叠TOM,基于TOM构建基因间的分层聚类树,动态剪切树法合并MEs相似度较高的模块,最终把基因聚类成3个模块,即Turquoise模块(2345个基因)、Blue模块(220个基因)、Grey模块(3个基因)(图2A),其中将不能聚类到任何模块的基因归于Grey模块,在后续分析中将其移除。进一步的共表达模块与临床表型的相关性热图分析(图2B)显示,Turquoise模块与OSA相关性最强(r=-0.98,P=0.000),以其作为关键模块进行GS与MM分析,Turquoise模块内基因与临床表型相关性良好,呈明显线性相关(图2C),分布在右上角的基因既与其他基因关联性高又与OSA的发病有密切联系,有助于疾病关键基因的识别。因此,笔者用STRING数据库对Turquoise模块构建蛋白相互作用网络,并用Cytoscape软件可视化节点度最高的前30个基因,结果发现,该模块存在多个枢纽基因,如SLC2A2、PRL、SST等(图2D)。
3.关键模块的功能富集分析:利用Cytoscape中的ClueGo插件对Turquoise模块内基因进行GO和KEGG富集分析,结果以P≤0.01为入选标准。该模块的基因功能主要注释于GO:0004984嗅觉受体活性,GO:0043227膜结合细胞器,GO:0005654核质,GO:0045184蛋白质定位。另外,KEGG通路分析显示该模块基因主要富集于嗅觉转导通路(hsa04740)和神经活性配体-受体相互作用通路(hsa04080)(图3)。
4.关键基因的识别与验证:从Top30基因构建的PPI网络中,依据节点度值选取排名前3位的基因SLC2A2、PRL、SST作为后续分析与验证的关键基因。笔者整合GSE135917和GSE38792数据集的样本(包括20例OSA组织和16例正常组织),通过分析各关键基因的表达水平发现,与正常组织比较,SLC2A2、PRL、SST在OSA组织中的表达明显降低(P均<0.01),与芯片分析结果一致(图4A)。随后,进行ROC分析以确定3个关键基因的诊断价值及在基因表达水平预测OSA的最佳截断值(图4B,表1),结果提示这3个关键基因很可能与OSA的发生、发展有重要联系。
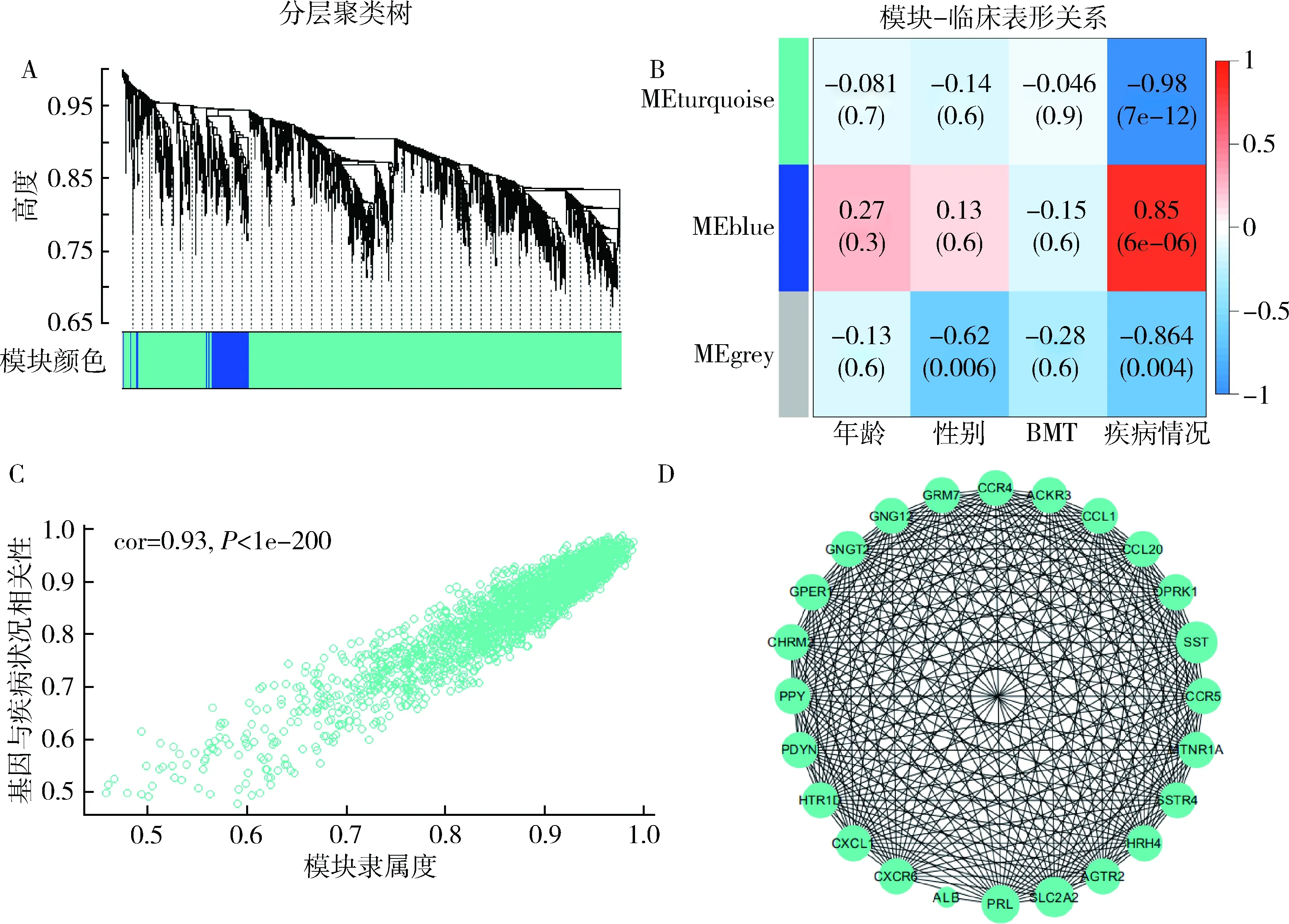
图2 加权基因共表达网络的构建 A.分层聚类树与共表达模块;B.模块与临床表型的相关性热图;C.Turquoise模块内基因与临床表型数据关联性; D.Turquoise模块内节点度最高的30个基因构建的蛋白相互作用网络图
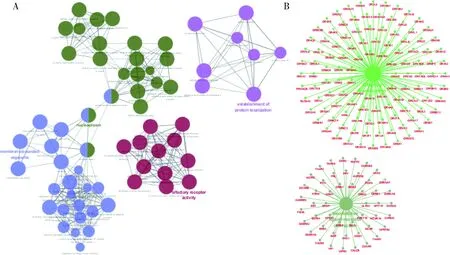
图3 Turquoise模块的功能富集分析 A.GO富集分析;B.KEGG信号通路分析

图4 关键基因的验证 A.GSE135917与GSE38792中关键基因的表达水平;B.关键基因的ROC曲线

表1 关键基因的ROC曲线分析
讨 论
OSA发病机制复杂,涉及多个器官系统的病理生理变化[2]。既往研究多数从单个基因出发,仅能对生物学过程做出局部解释,WGCNA通过构建基因间的邻接矩阵、拓扑重叠TOM,识别高度协同变化的基因模块,并能结合临床信息,分析模块与临床表型的相关性,充分利用基因组大数据信息,对其进行整体全面探索[11]。本研究利用WGCNA法筛选与OSA显著相关的基因模块,并对模块内基因进行功能富集分析,这些基因主要与嗅觉转导、神经活性配体-受体相互作用等密切相关。随后对关键模块构建蛋白相互作用网络,将枢纽基因可视化后识别出3个关键基因,通过在另一张芯片上再次验证,发现SLC2A2、PRL、SST在OSA组织中表达均下调,进一步的ROC曲线分析显示这3个关键基因可能是OSA潜在的生物学标志物。目前3个基因在OSA中尚未有相关报道,但仍有证据提示它们与OSA存在潜在关联。
SLC2A2是溶质载体家族2成员,编码葡萄糖转运子样蛋白GLUT2,分布于肝、肾、肠、胰岛β细胞和中枢神经系统,促进葡萄糖在质膜上的被动转运,在控制机体葡萄糖稳态中起重要作用[15]。在芬兰糖尿病预防研究中,SLC2A2的单核苷酸多态性(SNPs)与糖耐量受损向2型糖尿病的转化相关,而且这种关联与体重变化无关[16]。Borglykke等[17]在评估46个2型糖尿病相关基因变异的单独效应和累积效应时发现,只有SCL2A2的小等位基因与心血管事件发生风险增加显著相关,而且这种关联与基线糖尿病状态无关。本研究结果表明,SLC2A2在OSA患者中的表达水平降低,且具有良好的识别能力(AUC=0.9594)。目前OSA对心血管疾病、代谢综合征和神经精神障碍的不利影响日益受到关注,一些基因变异参与OSA发病机制,且可能与OSA相关疾病的发生也存在因果关系[7]。因此,研究SLC2A2变异是否同时与OSA和共病疾病(如糖尿病、心血管疾病)相关,即SLC2A2变异可能通过不同的机制同时影响这两种表型,可能具有重要意义。
催乳素(PRL)是一种由脑垂体分泌的蛋白质激素,参与泌乳、生殖、血管生成、免疫反应和渗透调节等多种生物学过程[18]。PRL还可影响睡眠结构,PRL基因缺陷小鼠的快速眼动睡眠比野生型小鼠减少[19]。此外,在高催乳素血症患者中观察到代谢改变、体重易增加,这些患者在其催乳素正常化后体重可减轻[20]。PRL可通过调节LPL活性和脂肪生成,减少脂联素在人体脂肪组织中的释放来调节能量代谢[21]。在一项针对早发和病态肥胖的全基因组关联研究(GWAS)中发现,PRL基因附近的变异与常见肥胖和BMI变异相关[22]。Nilsson等[21]在芬兰西部的一项大规模人群研究中成功复制了这种关联,发现PRL基因附近的变异与男性肥胖的增加有关。本研究中,与正常组织比较,OSA患者PRL基因的表达显著降低。众所周知,肥胖使OSA的发病风险增加10~14倍,有望解释高达40%的AHI变异,鉴定决定“中间表型”的基因可能有助于识别OSA的易感基因[7]。因此,有必要进一步研究PRL变异是否通过睡眠结构改变或中间表型(如肥胖)导致OSA。
生长抑素(SST)是一种环肽激素,影响生长激素的释放和胃肠功能。SST可通过调节胃肠运动、肠道养分吸收和能量平衡来影响机体生长和体重[23]。此外,作为中枢神经系统中广泛存在的神经递质或调质,SST在突触可塑性和神经元活性的微调中发挥作用[24]。研究表明,SST+/nNOS+神经元功能障碍可能导致与慢波活动产生障碍和认知受损相关的病理生理变化,包括阿尔茨海默病(AD)、癫痫、精神分裂症和创伤性脑损伤等[25]。衰老早期大脑中SST表达下调导致脑啡肽酶活性逐渐下降,导致AD患者Aβ-淀粉样蛋白的沉积,因此,SST基因变异可能改变生长抑素的表达或功能,从而参与AD的发病过程[26]。本研究中SST在OSA样本中的表达降低,如上所述,OSA与心脑血管病、神经精神障碍等密切相关,一些基因变异与OSA和相关疾病的发生可能均存在因果关系,故SST变异是否会通过不同的机制效应同时导致OSA和认知功能障碍,有必要进一步验证。
综上所述,本研究通过WGCNA方法在OSA患者皮下脂肪组织中发现了2个共表达模块和3个关键基因,有助于为OSA和相关疾病的研究及其诊断治疗、靶点选择提供新的线索。但关键基因在OSA发生中的具体作用,仍需要开展进一步的体内、体外实验予以深入探讨。
