Uhrf1对肠上皮发育的影响
2021-02-02王芯悦李亮段秋慧李大力陈金联
王芯悦,李亮,段秋慧,李大力,陈金联
Uhrf1对肠上皮发育的影响
王芯悦1,3,李亮2,3,段秋慧2,李大力2,陈金联3
1. 安徽理工大学医学院,淮南 232000 2. 华东师范大学生命科学院,上海市调控生物学重点实验室,上海 200241 3. 上海交通大学附属第六人民医院南院消化内科,上海 201499
作为一种常见的表观遗传修饰类型,DNA甲基化对哺乳动物发育起着重要作用。Uhrf1作为重要的表观遗传调控因子,在DNA合成过程中可结合半甲基化的DNA同时招募DNA甲基转移酶1参与DNA甲基化的维持,保证遗传信息在细胞分裂前后的稳定传递。目前关于Uhrf1介导的DNA甲基化是否影响肠上皮发育过程尚不清楚。为探索Uhrf1在肠上皮发育中的作用,本研究成功构建了肠上皮特异性敲除的小鼠模型,利用HE染色对肠上皮组织形态学观察发现,与正常小鼠相比,敲除的小鼠肠上皮发育异常,主要表现为绒毛变短,数量减少,隐窝萎缩;通过表型分析发现,在小鼠肠上皮中特异性敲除后,细胞增殖明显受到抑制、凋亡细胞增加、细胞分化异常,同时肠干细胞相关基因表达降低。进一步对可能的分子机制进行初步探索发现Uhrf1缺失后 DNA甲基化水平大幅下降,诱发DNA损伤。本研究结果表明Uhrf1介导的DNA甲基化对肠上皮的正常发育成熟具有重要作用,有望丰富Uhrf1介导的DNA甲基化在体内的生物学功能,并为进一步明确Uhrf1介导的表观遗传调控机制提供实验依据。
DNA甲基化;Uhrf1;肠上皮发育
DNA甲基化作为一种动态的和可逆的表观遗传修饰,是由甲基转移酶(DNA methyltransferases, DNMTs)介导的对胞嘧啶的第5位碳原子进行甲基修饰,形成5-甲基胞嘧啶(5-methylcytosine, 5mC),主要发生在CpG二核苷酸位点上[1]。DNA甲基化在个体发育、基因表达调控及基因组稳定性等多种生物学过程中发挥重要作用[2~5]。
肠上皮作为人体更新较快的组织,上皮细胞的正常增殖与分化无论是在空间还是时间上都受到严格调控,增殖与分化间的失衡将会破坏上皮的完整性及其屏障功能,甚至引发肿瘤的形成。肠上皮细胞这种快速的更新速率与不同类型细胞自身的表观遗传状态密切相关,目前关于DNA甲基化如何调控肠上皮的早期发育和稳态建立仍不是很清楚。
UHRF1 (ubiquitin-like with PHD and RING finger domains 1)是泛素样含植物同源化结构域(plant homeodomain domain, PHD)和环指域蛋白家族的主要成员之一,对DNA甲基化的维持至关重要[6]。UHRF1作为一种多结构域蛋白,SRA结构域(set and ring associated domain)可以识别半甲基化的DNA,并招募DNMT1到复制叉处,保证在细胞复制时遗传信息由亲代向子代的稳定传递[7];PHD及Tudor结构域与甲基化组蛋白H3K9特异性地结合,参与异染色质的形成和维持,同时有利于DNMT1的正确定位,将组蛋白修饰与DNA甲基化紧密联系起来[8,9]。此外UHRF1的RING(really interesting new gene)结构域被证明是一种泛素连接酶,可以催化组蛋白H3K23等位点发生泛素化修饰,并被DNMT1所识别,促进DNMT1招募进而参与DNA甲基化的维持[10,11]。除了参与DNA甲基化外,UHRF1还与细胞周期调控及DNA损伤修复等诸多生物学过程相关[12,13]。随着UHRF1生物化学功能的不断阐明,关于由UHRF1介导的DNA甲基化在体内的生物学功能也得到研究学者的广泛关注。研究发现Uhrf1决定卵母细胞质量,影响卵母细胞成熟,对胚胎着床前的发育至关重要[14];在斑马鱼()中,Uhrf1的突变导致整体DNA甲基化水平降低,肝脏体积缩小,组织发育不全[15];在对胸腺细胞的研究中表明,敲除造成胸腺细胞数量明显降低,Uhrf1介导的表观遗传调控对胸腺细胞的正常发育必不可少[16];而在大脑皮层的研究中却发现Uhrf1的缺失对早期发育作用较小,不影响细胞的增殖[17]。为探讨Uhrf1及其介导的DNA甲基化在肠上皮发育中的作用,本研究构建了在肠上皮中特异性敲除的小鼠模型,并对表型及可能的分子机制进行分析,初步揭示了Uhrf1介导的DNA甲基化在肠组织系统中的功能。
1 材料与方法
1.1 实验动物
实验所用的小鼠遗传背景均C57BL/6J品系,饲养于华东师范大学实验动物中心SPF级清洁实验动物房。动物实验的设计与操作均符合华东师范大学动物伦理委员会相关规定并被授权,严格遵守实验动物3R原则。
1.2 实验试剂
苏木精和伊红染液购自南京建成生物工程研究所;免疫组化二抗试剂盒和DAB显色试剂盒购自德国VECTOR公司;免疫组化及苏木精–伊红染色相关的无水酒精、二甲苯、甲醇等购自上海国药集团;EB染液、dNTP混合物、ExTaq DNA聚合酶和6×DNA loading buffer等购自北京天根生化科技有限公司;RNA反转录试剂盒和Trizol等购自日本TaKaRa 公司;SYBR green购自上海翊圣生物科技有限公司;DNA marker购自美国Thermo Fisher公司;Ki67抗体(RM9106,美国Thermo Fisher公司);Cleaved Caspase3抗体 (9664#,美国Cell SignalingTechnology公司);Muc2 (Mucin 2)抗体(美国Santa Cruz Biotechnology公司);Dclk1(Doublecortin-like kinase 1)抗体(ab31704,英国Abcam),ChgA (Chromogranin A)抗体(ab15160,英国Abcam);Uhrf1抗体(61341,美国Active Motif公司);5mC抗体(39769,美国Active Motif公司);γH2AX (phosphorylation of histone H2AX)抗体(05636#,美国Millipore公司)。
1.3 Uhrf1条件性敲除小鼠的构建
通过基因打靶技术,在基因的第3号外显子和第4号外显子之间的内含子区域插入可以表达β-半乳糖苷酶(LacZ)和neo抗性蛋白以及polyA,同时两端还有2个同向的FRT (short flippase recognition target)序列,另外在第4号外显子两侧插入了同向的loxp序列。通过移植该ES细胞到假孕小鼠的子宫,获得可以条件性敲除的小鼠。取-LacZ小鼠与Flp工具鼠交配,Flp(flippase)重组酶发挥作用,切除FRT之间序列,得到仅在第4号外显子两侧插入同向的loxp序列的小鼠,使用此小鼠与在肠上皮中特异性表达重组酶Cre的工具鼠杂交,切割的第4号外显子,导致Uhrf1蛋白的翻译出现移码,从而敲除基因。
1.4 聚合酶式反应(polymerase chainreaction, PCR)
剪取小鼠脚趾并标记放入1.5 mL无菌EP管中,按1:500比例加入蛋白酶K与消化液,置于55度水浴锅中消化过夜,次日100度煮沸5分钟后离心,进行PCR扩增。引物由上海铂尚生物技术有限公司合成,序列Uhrf1(F):5ʹ-ACTCTTGATCTGTGCCCTGC-3ʹ和Uhrf1(R):5ʹ-ATCCCAGGCCTCCATACACT-3ʹ。扩增体系(总体积为20 μL):2 μL 10×buffer,2 μL dNTP混合物,2 μL引物,3 μL模板,0.2 μL Easy Taq酶,11 μL ddH2O。反应条件:95℃ 5 min;95℃ 30 s,60℃ 30 s,72℃ 40 s;循环35次。扩增后进行凝胶电泳,凝胶自动成像仪记录电泳图像,比对条带大小,确定小鼠基因型。
1.5 肠组织切片及苏木精–伊红染色(hematoxylin-eosin staining, HE)
采用颈椎脱臼法将小鼠处死后,分离小鼠肠组织,制成“瑞士卷”,用4%多聚甲醛固定过夜,包埋后将蜡块放入–20℃冰箱中保存,包埋好的组织制成5 μm切片,62℃烤片2 h,待干燥后进行脱蜡复水,具体过程为二甲苯I:7 min→二甲苯II:7 min→二甲苯:乙醇(1∶1):7 min→100%乙醇:4 min→95%乙醇:4 min→85%乙醇:4 min→75%乙醇:4 min→纯水:3 min→苏木精:5 min(具体视苏木精浓度而定)→1%盐酸酒精分色:10~30 s→蒸馏水中返蓝:15 min→伊红染色:30 s~1min→脱水及中性树脂封片→显微镜下拍照。
1.6 免疫组织化学染色
组织切片脱蜡复水同HE染色→3%的过氧化氢避光处理13 min→抗原修复液置于100℃处理20 min→PBST润洗3次,每次5 min→封闭液室温封闭30 min以上→滴加一抗于4℃孵育过夜→回收一抗,PBST润洗3次→滴加HRP室温避光反应30 min→PBST润洗3次→显色3~5 min,深度适宜后蒸馏水中终止显色反应→苏木精复染→盐酸酒精分色→自来水中返蓝15 min→脱水封片同HE染色。
1.7 实时荧光定量PCR (quantitative real-time PCR, qRT-PCR)分析
小鼠脱颈椎处死后取出肠组织液氮研磨后加入1 mLTrizol,按RNA抽提试剂盒说明书提取总 RNA,利用反转录试剂盒合成cDNA。qRT-PCR扩增体系为25 μL,包括:12.5 μL (2×) SYBR Premix ExTaq,2 μL引物,1 μL cDNA,9.5 μL RNase FreeH2O。扩增条件:95℃ 5 min;95℃ 30 s,60℃ 30 s,72℃ 30 s,共40个循环,每个样进行3次重复。根据每个样品与内参基因所得的Ct值,利用2–ΔΔCt公式分析目的基因相对表达量。基因的扩增引物序列见表1。

表1 qRT-PCR引物序列
1.8 统计方法
用Graphpad Prism 6软件进行统计学分析,各组之间的比较用平均数±标准差表示,<0.05被认为有统计学意义。
2 结果与分析
2.1 Uhrf1基因条件性敲除小鼠的构建与鉴定
Uhrf1小鼠是以基因的第4号外显子为靶基因,在其两端插入loxp位点,与表达Cre酶的工具鼠交配后获得VillinCre-Uhrf1小鼠,从而切除相同方向的两个loxp位点间第4号外显子序列,导致UHRF1蛋白翻译时出现移码,从而实现靶基因的敲除(图1A)。设计引物针对loxp序列进行PCR,对子代进行基因型鉴定。如图1B所示,仅扩增出305 bp条带者为Uhrf1,扩增出305 bp和106 bp两条带的为Uhrf1,仅扩增出106 bp条带者为Uhrf1。通过对子代小鼠进行基因型的鉴定,观察发现VillinCre-Uhrf1小鼠可以正常出生,但是出生率低于理论预期,部分小鼠在断奶前后出现显著死亡情况,很难发育到成年阶段,因此后续实验选取出生后3周小鼠作为实验研究对象。
2.2 Uhrf1基因条件性敲除小鼠的敲除效果
为了探索Uhrf1在肠发育及稳态建立中的作用,首先采用免疫组织化学染色法对Uhrf1表达量及位置进行检测,如图2A(左)染色结果显示,着色区域阳性细胞仅定位于肠隐窝部位,肠的其他部位未见有表达,说明Uhrf1主要存在于肠隐窝增殖性细胞中,即主要包含小肠干细胞及快速增殖的祖细胞。进一步采用Cre-loxp重组技术构建肠上皮中特异性敲除的突变小鼠,并采用免疫组织化学染色和qRT-PCR方法检测Uhrf1在肠组织中的表达情况,免疫组织化学技术结果显示在条件性敲除小鼠的肠隐窝底部区域并未检测到Uhrf1的表达(图2A,右);由图2B结果可见,基因敲除小鼠肠组织中mRNA表达水平低于对照组小鼠。结果表明Cre重组酶可以发挥正常功能,导致基因敲除,证实基因条件性敲除小鼠构建成功。
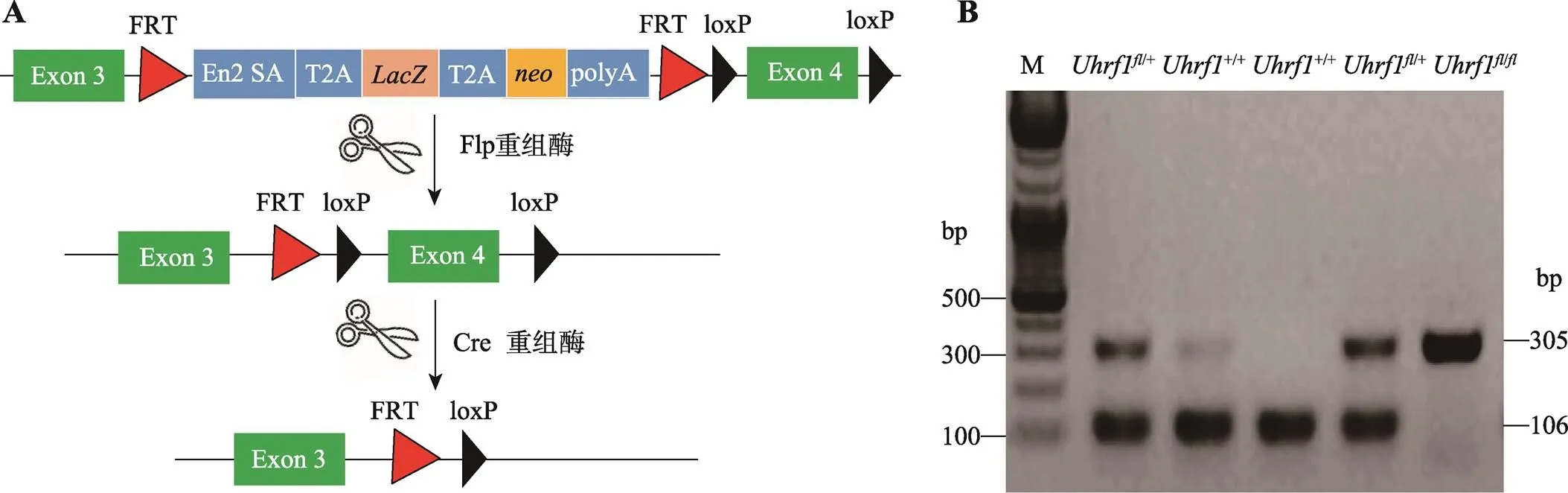
图1 Uhrf1基因条件性敲除小鼠的构建与鉴定
A:基因条件性敲除小鼠构建示意图。B:PCR鉴定基因敲除结果。M:DNA marker。
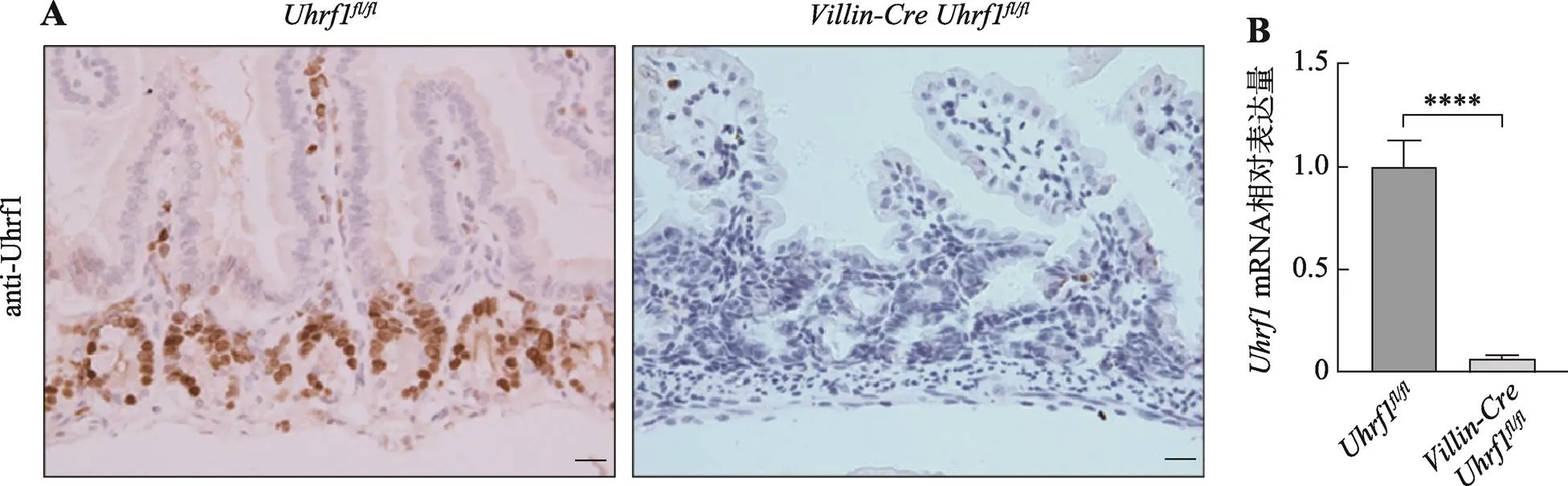
图2 Uhrf1在肠组织中的表达情况
A:野生型和突变型小鼠肠组织中Uhrf1抗体免疫组织化学染色。标尺:20 μm。B:野生型与突变型小鼠肠组织中mRNA表达量对比。****:<0.0001。
2.3 敲除Uhrf1造成肠上皮组织形态异常
为探究Uhrf1对肠上皮组织形态学的影响,选取出生后3周的野生型与敲除小鼠,脱颈椎处死后取小肠组织制作石蜡切片,通过HE染色进行组织形态学分析发现,与对照组小鼠相比(图3A),Uhrf1的缺失导致肠绒毛长度明显变短,隐窝发生萎缩,同时两者数量减少,表明敲除后肠上皮发育过程受阻(图3B),肠上皮组织形态的建立依赖于Uhrf1。
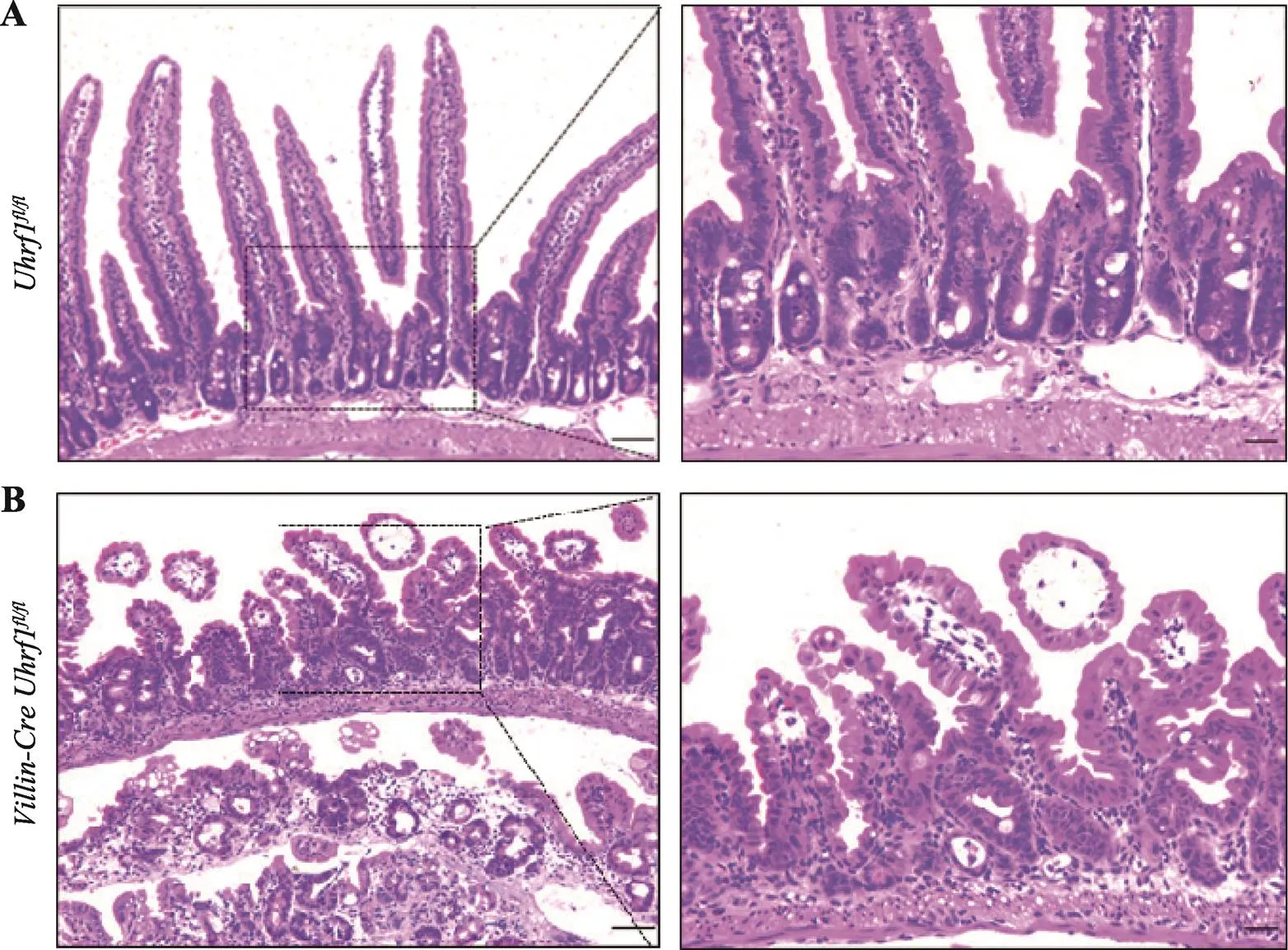
图3 野生型及突变型小鼠肠组织HE染色
A:野生型小鼠肠组织切片观察。B:突变型小鼠肠组织切片观察。右图均是左图虚线区域的放大示意图。标尺左:50 μm,标尺右:20 μm。
2.4 Uhrf1的缺失影响肠上皮细胞的增殖与分化能力,诱导细胞凋亡
隐窝作为肠上皮的功能单位,负责维持细胞增殖与分化的平衡。Uhrf1特异性表达隐窝底部区域,暗示其可能影响上皮细胞的增殖和分化。因此选取3周龄VillinCre-Uhrf1小鼠,同时选取同窝Uhrf1的小鼠作为对照,采用免疫组织化学染色方法检测肠上皮细胞的增殖、凋亡和分化情况,并进一步对凋亡细胞及各类分化细胞数量进行检测,在20倍显微镜视野下拍照,随机选取3个形态相似视野,统计每个视野阳性细胞数。结果显示:与对照组小鼠相比,胚胎期敲除导致增殖标记蛋白Ki67阳性细胞数量显著减少(图4A),说明肠上皮细胞的正常增殖受到明显抑制。而凋亡相关蛋白Caspase3阳性细胞数量明显增加,并且主要集中在隐窝底部区域(图4,B和F),表明Uhrf1的缺失不仅影响了细胞增殖,而且促使肠上皮细胞发生凋亡。敲除后肠上皮细胞增殖受到抑制以及凋亡增加暗示分化过程可能也受到影响,因此为进一步了解Uhrf1对肠上皮分化细胞的影响,对杯状细胞-Muc2、内分泌细胞-ChgA、Tuft细胞-Dclk1标记阳性细胞数目进行检测,结果发现VillinCre-Uhrf1小鼠肠组织Muc2 (图4,C和G)、ChgA (图4,D和H)和Dclk1 (图4,E和I)阳性细胞数减少,表达下降,说明敲除Uhrf1明显影响了肠上皮细胞的正常分化过程。因此,这些研究结果表明Uhrf1是调节肠上皮细胞增殖与分化的关键因子。

图4 Uhrf1fl/fl和VillinCre-Uhrf1fl/fl小鼠肠组织中的表型分析
A:肠组织增殖标记蛋白Ki67免疫染色。B:肠组织凋亡标志蛋白Caspase3-cleaved免疫染色。红色箭头指示凋亡阳性细胞,左上角为红色箭头区域放大图。C:杯状细胞标记蛋白Muc2免疫染色。D:内分泌细胞标记蛋白ChgA免疫染色。E:Tuft细胞标记蛋白Dclk1免疫染色。标尺:50 μm F~I:野生型与突变型小鼠Caspase3-cleaved、Muc2、ChgA、Dclk1阳性细胞统计结果。*:<0.05;**:<0.01;***:<0.001。**:<0.01;***:<0.001;****:<0.0001。
2.5 敲除Uhrf1肠干细胞标志基因的表达降低
Lgr5+肠干细胞作为一种快速增殖的细胞,对肠组织生理功能及稳态的维持起着重要的作用,因此为了解基因敲除鼠中Lgr5+干细胞有没有受到影响,本研究进一步在分子水平上对Lgr5+干细胞相关的基因:、、和表达情况进行检测,结果发现,与对照组小鼠相比,在肠组织中特异性敲除后,造成肠干细胞相关标志基因的表达降低(图5),表明Uhrf1可能对肠干细胞数量具有一定的影响。
2.6 敲除Uhrf1导致DNA甲基化水平下降,诱导DNA损伤
如前所述,UHRF1作为表观遗传调控因子,在DNA甲基化维持中起着关键作用。为确定Uhrf1的缺失是否会导致肠上皮基因组甲基化的丢失,采用免疫组织化学染色方法对5mC表达水平进行检测发现,与对照组小鼠相比,条件性敲除小鼠的肠组织中5mC水平明显降低,表明敲除会造成肠上皮组织甲基化水平显著下降(图5A)。研究发现DNA甲基化水平降低会引起DNA损伤应答反应,因此对DNA损伤标志物γH2AX进行免疫组织化学染色,发现敲除后在肠隐窝部位可以看到明显的DNA损伤(图5B),说明Uhrf1的缺失导致肠上皮组织DNA损伤增加。

图5 肠干细胞相关标志基因的表达
3 讨论
DNA甲基化作为最稳定的表观遗传修饰,对体内多个自我更新组织中基因的表达调控至关重要,DNA甲基化的异常将造成基因表达的改变,从而导致疾病的发生[18,19]。多项研究证明Uhrf1可招募Dnmt1,在增殖性细胞中维持DNA甲基化信息在细胞分裂前后的稳定传递,缺失Uhrf1将会导致细胞内的DNA甲基化水平大幅下降[20]。本研究发现Uhrf1主要表达在肠隐窝底部区域主要是肠干细胞和快速增殖的祖细胞,而在绒毛等其他部位未见有表达,进一步证明Uhrf1在增殖性细胞中表达丰富,这与之前报道的Uhrf1在其他组织系统中的表达相一致[21]。
研究表明Uhrf1可以影响不同组织细胞的增殖和分化并与凋亡过程相关。在对结肠调节性T细胞的研究中发现Uhrf1缺陷的小鼠表现为细胞增殖和正常发育成熟过程受阻,免疫功能降低,并自发形成结肠炎[22]。在斑马鱼肝脏中,敲减可显著抑制肝细胞增殖,造成凋亡增加[23]。在四肢间充质细胞中敲除后,由于破坏了软骨细胞增殖及分化过程,造成小鼠肢体明显缩短[24]。同样,本研究结果发现Uhrf1的缺失抑制了肠上皮细胞的增殖与分化,诱导细胞凋亡,导致肠上皮发育异常, 并造成肠干细胞相关标记基因表达水平的降低。已有研究证明DNA甲基化水平的降低将会导致基因组不稳定,突变率增加,从而引起DNA损伤反应[25~27]。Amy等[28]在结肠癌细胞系HCT116中敲减发现,Uhrf1的丢失导致DNA损伤反应的激活,主要表现为组蛋白H2AX在第139位丝氨酸的磷酸化以及细胞周期检测点激酶2 (checkpoint kinase 2, CHK2)第68位苏氨酸的磷酸化等,并造成细胞通过caspases 8和3途径发生凋亡。为进一步探索基因是如何诱发细胞凋亡,通过免疫组织化学染色方法对肠上皮组织甲基化水平及DNA损伤标志物γH2AX进行检测,结果显示与对照组小鼠相比,Uhrf1的缺失导致肠上皮组织整体甲基化水平降低,DNA损伤增加,因此初步推测可能是Uhrf1的缺失造成DNA甲基化水平大幅下降,诱发DNA损伤,进而引起肠上皮细胞增殖分化减缓,凋亡增加。目前已有研究表明DNA甲基化可以通过调控发育过程中细胞增殖和分化间的平衡来调节肠上皮稳态的建立。Sheaffer等[29~31]研究发现Dnmt1介导的维持性DNA甲基化对肠上皮细胞分化过程中的基因表达调控起着关键作用,胚胎期敲除发现Dnmt1的缺失导致小鼠肠上皮细胞增殖下降、绒毛数量减少、基因组甲基化水平降低,并采用转录组学测序技术分析发现与DNA损伤及细胞周期相关的基因表达升高,造成细胞周期阻滞,从而引起细胞死亡,这与本研究中在小鼠胚胎期肠上皮细胞中特异性敲除的表型相类似;进一步在成体小鼠中的研究发现,尽管短期内敲除可导致小鼠体重下降,基因组稳定性及甲基化水平降低,但两个月左右由于Dnmt3b的激活,DNA甲基化水平及肠上皮完整性可以逐渐恢复到正常水平,Dnmt1和Dnmt3b共同负责成体小鼠肠上皮甲基化的维持,目前关于Uhrf1在成体小鼠肠组织中的功能还有待建立动物模型进行下一步的研究。据报道Uhrf1可以通过靶向多个信号通路发挥生物学功能。例如Chen等[32]证明Uhrf1可以通过影响细胞周期抑制蛋白CDKN1A (cyclin dependent kinase inhibitor 1A)以及调节B细胞增殖和衰老家族的()表达水平来促进B细胞增殖,进一步应用亚硫酸盐测序分析发现基因的CpG位点的DNA甲基化水平在敲除后明显降低。Xiang 等[33]发现Uhrf1的缺失导致基底细胞阻滞在G1期,损害了气道再生过程,并表明Uhrf1可能影响了与G1/S期转变相关的细胞周期蛋白依赖性激酶的活性。在自然杀伤性T细胞中Uhrf1被证明可以通过调节蛋白激酶B/哺乳动物雷帕霉素白蛋白(protein kinase B/mammalian target of rapamycin, Akt-mTOR)信号轴来控制细胞的分化,影响细胞的发育[34]。目前关于Uhrf1介导的DNA甲基化对肠上皮发育具体调控机制尚不清楚,考虑到Uhrf1和Dnmt1在甲基化维持中的作用,Uhrf1和Dnmt1是否具有类似的功能与机制,需要更进一步的探索。

图6 Uhrf1介导的DNA甲基化对肠上皮细胞影响
A:野生型与条件性敲除Uhrf1小鼠肠上皮中5mC免疫组织化学染色。B:野生型与条件性敲除Uhrf1小鼠肠上皮中DNA损伤标志物γH2AX 免疫组织化学染色。红色箭头表示DNA损伤阳性细胞。标尺:50 μm。
综上所述,本研究通过构建在肠上皮中特异性敲除的小鼠模型,发现在胚胎期敲除后,肠上皮发育异常,上皮稳态失衡,Uhrf1的缺失导致DNA甲基化的维持过程被破坏,诱发DNA损伤反应,造成肠上皮细胞的增殖与分化受阻,细胞凋亡增加,首次揭示了Uhrf1介导的DNA甲基化在肠上皮发育中的重要作用,为进一步明确Uhrf1介导的表观遗传机制提供了实验依据。
[1] Smith ZD, Meissner A. DNA methylation: roles in mammalian development., 2013, 14(3): 204–220.
[2] Greenberg MVC, Bourc'his D. The diverse roles of DNA methylation in mammalian development and disease., 2019, 20(10): 590–607.
[3] Schübeler D. Function and information content of DNA methylation., 2015, 517(7534): 321–326.
[4] Smith ZD, Sindhu C, Meissner A. Molecular features of cellular reprogramming and development., 2016, 17(3): 139–154.
[5] Newkirk SJ, An WF. Uhrf1: a jack of all trades, and a master epigenetic regulator during spermatogenesis., 2020, 102(6): 1147–1152.
[6] Xue BS, Zhao JS, Feng PH, Xing J, Wu HL, Li Y. Epigenetic mechanism and target therapy of Uhrf1 protein complex in malignancies., 2019, 12: 549–559.
[7] Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA, Shinga J, Mizutani-Koseki Y, Toyoda T, Okamura K, Tajima S, Mitsuya K, Okano M, Koseki H. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA., 2007, 450(7171): 908–912.
[8] Xie S, Qian CM. The growing complexity of Uhrf1- mediated maintenance DNA methylation., 2018, 9(12): 600.
[9] Cheng JD, Yang Y, Fang J, Xiao JX, Zhu TT, Chen F, Wang P, Li Z, Yang HR, Xu YH. Structural insight into coordinated recognition of trimethylated histone H3 lysine 9 (H3K9me3) by the plant homeodomain (PHD) and tandem tudor domain (TTD) of Uhrf1 (ubiquitin-like, containing PHD and RING finger domains, 1) protein., 2013, 288(2): 1329–1339.
[10] Nishiyama A, Yamaguchi L, Sharif J, Johmura Y, Kawamura T, Nakanishi K, Shimamura S, Arita K, Kodama T, Ishikawa F, Koseki H, Nakanishi M. Uhrf1- dependent H3K23 ubiquitylation couples maintenance DNA methylation and replication., 2013, 502(7470): 249–253.
[11] Harrison JS, Cornett EM, Goldfarb D, DaRosa PA, Li ZM, Yan F, Dickson BM, Guo AH, Cantu DV, Kaustov L, Brown PJ, Arrowsmith CH, Erie DA, Major MB, Klevit RE, Krajewski K, Kuhlman B, Strahl BD, Rothbart SB. Hemi-methylated DNA regulates DNA methylation inheritance through allosteric activation of H3 ubiquitylation by Uhrf1., 2016, 5: e17101.
[12] Jeanblanc M, Mousli M, Hopfner R, Bathami K, Martinet N, Abbady AQ, Siffert JC, Mathieu E, Muller CD, Bronner C. The retinoblastoma gene and its product are targeted by ICBP90: a key mechanism in the G1/S transition during the cell cycle.2005, 24(49): 7337–7345.
[13] Tian YY, Paramasivam M, Ghosal G, Chen D, Shen X, Huang YL, Akhter S, Legerski R, Chen JJ, Seidman MM, Qin J, Li L. Uhrf1 contributes to DNA damage repair as a lesion recognition factor and nuclease scaffold., 2015, 10(12): 1957–1966.
[14] Maenohara S, Unoki M, Toh H, Ohishi H, Sharif J, Koseki H, Sasaki H. Role of Uhrf1 in de novo DNA methylation in oocytes and maintenance methylation in preimplantation embryos., 2017, 13(10): e1007042.
[15] Jacob V, Chernyavskaya Y, Chen XT, Tan PS, Kent B, Hoshida Y, Sadler KC. DNA hypomethylation induces a DNA replication-associated cell cycle arrest to block hepatic outgrowth in Uhrf1 mutant zebrafish embryos.,2015, 142(3): 510–521.
[16] Zhang YW, Chen YS, Ma R, Jiang YW, Liu J, Lin YT, Chen SQ, Xia MY, Zou F, Zhang JS, Pan T, Wang L, Wei L, Zhang H. Uhrf1 controls thymocyte fate decisions through the epigenetic regulation of EGR1 expression., 2020, 204(12): 3248–3261.
[17] Ramesh V, Bayam E, Cernilogar FM, Bonapace IM, Schulze M, Riemenschneider MJ, Schotta G, Götz M. Loss of Uhrf1 in neural stem cells leads to activation of retroviral elements and delayed neurodegeneration.,2016, 30(19): 2199–2212.
[18] Sen GL, Reuter JA, Webster DE, Zhu L, Khavari PA. DNMT1 maintains progenitor function in self-renewing somatic tissue., 2010, 463(7280): 563–567.
[19] Yang XD, Han W, Liu F. DNA methylation in vertebrate embryogenesis.(, 2012, 34(9): 1108– 1113.杨晓丹, 韩威, 刘峰. DNA甲基化与脊椎动物胚胎发育. 遗传, 2012, 34(9): 1108–1113.
[20] Bostick M, Kim JK, Estève PO, Clark A, Pradhan S, Jacobsen SE. Uhrf1 plays a role in maintaining DNA methylation in mammalian cells., 2007, 317(5845): 1760–1764.
[21] Blanchart A, Navis AC, Assaife-Lopes N, Usoskin D, Aranda S, Sontheimer J, Ernfors P. Uhrf1 licensed self-renewal of active adult neural stem cells., 2018, 36(11): 1736–1751.
[22] Obata Y, Furusawa Y, Endo TA, Sharif J, Takahashi D, Atarashi K, Nakayama M, Onawa S, Fujimura Y, Takahashi M, Ikawa T, Otsubo T, Kawamura YI, Dohi T, Tajima S, Masumoto H, Ohara O, Honda K, Hori S, Ohno H, Koseki H, Hase K. The epigenetic regulator Uhrf1 facilitates the proliferation and maturation of colonic regulatory T cells., 2014, 15(6): 571–579.
[23] Sadler KC, Krahn KN, Gaur NA, Ukomadu C. Liver growth in the embryo and during liver regeneration in zebrafish requires the cell cycle regulator, Uhrf1., 2007, 104(5): 1570–1575.
[24] Yamashita M, Inoue K, Saeki N, Ideta-Otsuka M, Yanagihara Y, Sawada Y, Sakakibara I, Lee J, Ichikawa K, Kamei Y, Iimura T, Igarashi K, Takada Y, Imai Y. Uhrf1 is indispensable for normal limb growth by regulating chondrocyte differentiation through specific gene expression., 2018, 145(1): dev157412.
[25] Jenkins Y, Markovtsov V, Lang W, Sharma P, Pearsall D, Warner J, Franci C, Huang B, Huang JN, Yam GC, Vistan JP, Pali E, Vialard J, Janicot M, Lorens JB, Payan DG, Hitoshi Y. Critical role of the ubiquitin ligase activity of Uhrf1, a nuclear RING finger protein, in tumor cell growth., 2005, 16(12): 5621–5629.
[26] Ma HH, Chen H, Guo X, Wang ZT, Sowa ME, Zheng LJ, Hu SB, Zeng PY, Guo R, Diao JB, Lan F, Harper JW, Shi YG, Xu YH, Shi Y. M phase phosphorylation of the epigenetic regulator uhrf1 regulates its physical association with the deubiquitylase USP7 and stability., 2012, 109(13): 4828–4833.
[27] Loughery JEP, Dunne PD, O'Neill KM, Meehan RR, McDaid JR, Walsh CP. Dnmt1 deficiency triggers mismatch repair defects in human cells through depletion of repair protein levels in a process involving the DNA damage response.2011, 20(16): 3241– 3255.
[28] Tien AL, Senbanerjee S, Kulkarni A, Mudbhary R, Goudreau B, Ganesan S, Sadler KC, Ukomadu C. Uhrf1 depletion causes a G2/M arrest, activation of DNA damage response and apoptosis., 2011, 435(1): 175–185.
[29] Sheaffer KL, Kim R, Aoki R, Elliott EN, Schug J, Burger L,Schübeler D, Kaestner KH. DNA methylation is required for the control of stem cell differentiation in the small intestine., 2014, 28(6): 652–664.
[30] Elliott EN, Sheaffer KL, Schug J, Stappenbeck TS, Kaestner KH. Dnmt1 is essential to maintain progenitors in the perinatal intestinal epithelium., 2015, 142(12): 2163–2172.
[31] Elliott EN, Sheaffer KL, Kaestner KH. The 'de novo' DNA methyltransferase Dnmt3b compensates the Dnmt1-deficient intestinal epithelium., 2016, 5: e12975.
[32] Chen C, Zhai SL, Zhang L, Chen JJ, Long XH, Qin J, Li JH, Huo R, Wang XM. Uhrf1 regulates germinal center B cell expansion and affinity maturation to control viral infection., 2018, 215(5): 1437–1448.
[33] Xiang HD, Yuan LF, Gao X, Alexander PB, Lopez O, Lau C, Ding Y, Chong MY, Sun T, Chen R, Liu SQ, Wu HY, Wan Y, Randell SH, Li QJ, Wang XF. Uhrf1 is required for basal stem cell proliferation in response to airway injury., 2017, 3: 17019.
[34] Cui Y, Chen XF, Zhang JL, Sun X, Liu HF, Bai L, Xu CQ, Liu XL. Uhrf1 controls iNKT cell survival and differentiation through the Akt-mTOR axis., 2016, 15(2): 256–263.
Effect of Uhrf1 on intestinal development
Xinyue Wang1,3, Liang Li2,3, Qiuhui Duan2, Dali Li2, Jinlian Chen3
As a best-characterized epigenetic modification, DNA methylation plays an important role in mammalian development. Uhrf1 is a critical epigenetic regulator that can bind to hemimethylated DNA and recruit DNA methyltransferase 1 to maintain DNA methylation. So far, the role of Uhrf1-mediated DNA methylation in intestinal development is still unknown. In order to investigate the impact of Uhrf1 deletion in intestinal development, we have successfully constructed the epithelial-specificknockout mouse model. After Uhrf1 ablation, we found the mutant mice exhibited abnormal epithlial structure with less and shorter villi and shrinked crypts compared with wild type micehematoxylin-eosin staining.Further analysis showed that Uhrf1 deletion in the intestinal epithelium significantly decreased the cell proliferation and induced cell apoptosis. In addition, Uhrf1 deletion inhibited the normal epithelial differentiation and the expression of intestinal stem cell marker genes.Preliminary mechanism study revealed that loss of Uhrf1 caused global DNA hypomethylation which induced DNA damage in crypt cells.Taken together,our data suggested that DNA methylation mediated by Uhrf1 is vital forthe normal intestinal development.Our results enriched therole of Uhrf1 and laid the foundation for further epigeneticregulatory mechanismexploration.
DNA methylation;Uhrf1;intestinal development
2020-11-26;
2020-12-22
上海市自然科学基金项目(编号:16ZR1429100)资助[Supported by the National Natural Science Foundation of Shanghai(No. 16ZR1429100)]
王芯悦,在读硕士研究生,专业方向:消化内科学。E-mail: 1294008801@qq.com
李大力,博士,研究员,研究方向:基因编辑。E-mail: dlli@bio.ecnu.edu.cn陈金联,博士,主任医师,研究方向:消化道肿瘤与肝病。E-mail: wqq_021002@163.com
10.16288/j.yczz.20-337
2021/1/8 14:13:16
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20210107.1324.006.html
(责任编委: 赵冰)
