聚乙二醇二羧基醚合成工艺优化
2021-01-30张用芳米玉伟曾涑源孙德志
张用芳,米玉伟,刘 敏,,曾涑源,孙德志
(1.聊城大学 生物制药研究院,山东 聊城 252059;2.聊城大学 化学化工学院,山东 聊城 252059)
0 引言
水溶性聚合物修饰剂聚乙二醇(polyethylene glycol,PEG),又名α-氢-ω-羟基(氧-1,2-乙二基)聚合物、聚氧化乙烯(PEO-LS),是平均分子量在200-6000 u的乙二醇高聚物的总称,其结构式为HO(CH2CH2O)nH.随着平均分子量的增加,其外观逐渐从无色无臭粘稠液体转变为蜡状固体,分子量200-600 u者常温下是液体,分子量在600 u以上者就逐渐变为半固体状,分子量1000 u以上为固体,其吸湿能力相应降低。工业上,一般采用乙二醇在高温高压下聚合、环氧乙烷与水/乙二醇逐步加成聚合合成。
PEG是非离子型水溶性聚合物,PEG及其系列产品无毒、无刺激性,具有良好的两亲性,易溶于水和乙醇、氯仿、N,N-二甲基甲酰胺(DMF)、二氧六环等绝大多数有机溶剂,对热、酸、碱稳定,常用作药物赋形剂、增塑剂、乳化剂及润湿剂等[1-6]。PEG是美国食品药品管理局(FDA)批准的几种可用于药用合成的聚合物之一,PEG具有生物相容性良好、免疫原性低、对人体刺激不明显等优点[2,3,6-8],而且可以通过肾脏排出体外,在体内无积累[9,10]。
药物的PEG修饰是将活化的PEG经由化学方法偶联到药物上的过程,即PEG化(PEGylation)。活化的PEG常用来修饰酶、多肽、蛋白质[11-13]及小分子药物(如喜树碱、紫杉醇等[14-16])等非水溶性药物,用以克服此类药物的诸多缺点,如不良物理化学性质、半衰期短、稳定性差和毒性高等,同时保持他们的治疗效果。然而,由于PEG的端基羟基与其它基团发生反应时,极容易破坏被修饰的药物或其他物质,一般需要先对其进行活化。活化的PEG能够有效地改善药效及药物动力学等特性,进一步增加非水溶性药物的临床应用范围。
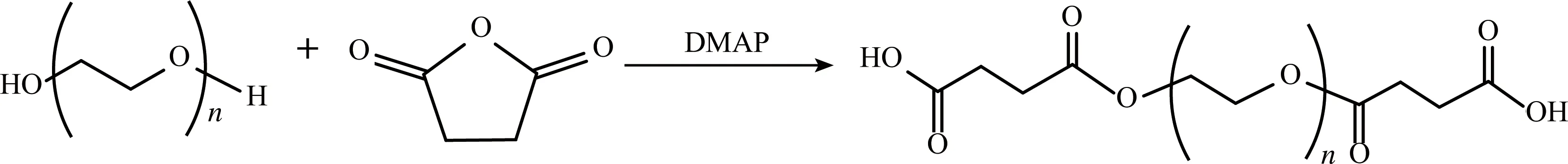
PEG的传统活化方法有氰脲酞氯法、烷基化、酰基化等,以上方法具有毒性大、温度高、反应条件苛刻等缺点[17-19]。本研究采用丁二酸酐(SA)对PEG进行活化,以低毒的DMF为溶剂,低温合成了平均分子量约2200 u的聚乙二醇二羧基醚(PEG-biCOOH)。核磁共振氢谱、红外光谱和质谱测试结果表明,在催化剂(4-二甲氨基吡啶,DMAP)的作用下,开环的SA与PEG端基羟基键合,成功合成目标产物PEG-biCOOH,且PEG分子链本身未引入其它基团,为其在非水溶性药物的修饰、改性、制备药物纳米颗粒及合成前体药物等研究打下了良好的基础。此研究系统考察了合成聚乙二醇二羧基醚的影响因素,反应溶剂为DMF时,探明其最优反应条件为PEG与SA的摩尔比1:2.8,反应时间24 h,反应温度60 ℃,所得产品收率为97.8%,经透析后收率为86.7%,高于文献值[2].此研究为以PEG为医药中间体合成前体药物等相关研究奠定了基础。
1 实验部分
1.1 试剂
聚乙二醇(polyethylene glycol,PEG,平均分子量2000 u),丁二酸酐(Succinic anhydride,SA,分子量100 u),二氧六环(Dioxane),N,N-二甲基甲酰胺(N,N-Dimethylformamide,DMF),甲苯(Methylbenzene),盐酸(Hydrochloric acid,HCl),二氯甲烷(Dichloromethane,CH2Cl2),乙醚(Ether),均为国药分析纯试剂,4-二甲氨基吡啶(4-Dimethylaminopyridine,DMAP,纯度99%)购于Alfa Aesar试剂公司。所有试剂未经进一步提纯。实验中使用的水为超纯水,其电阻率≥18.2 MΩ,采用Ultra-Pure 型超纯水系统(上海和泰仪器有限公司)制备。
1.2 聚乙二醇二羧基醚的合成
称取2 mmol PEG(4 g)和一定量的SA(4.4-8.4 mmol),溶于30 mL不同溶剂(分别是DMF、甲苯、二氧六环)中,再加入一定量的DMAP(用量与SA相同),于氮气氛围下,混合物反应不同的温度(25-100 ℃)和时间(3-48 h)。反应完成后,加入20 mL水,用5 mol/L HCl调节pH至2.5,用二氯甲烷萃取三次(50 mL/次),浓缩后加入过量的乙醚,使产物沉淀,将粗产物溶于少量水,之后用三次水透析(透析袋截留分子量1000 u)两天,除水浓缩后进行冷冻干燥(12 h),最终得白色絮状粉末,4 ℃下储存[20-24]。
实验得出的聚乙二醇二羧基醚(PEG-biCOOH)的最优反应条件为:以DMF为溶剂,PEG与SA(5.6 mmol)的摩尔比为1:2.8,反应温度为60 ℃,反应时间为24 h,此条件下得到的产品记为S1。具体反应条件和产品编号列于表1。每个实验平行3次,取平均值反应方程式如:
1.3 表征
以氘代DMSO-d6为溶剂,采用400 MHz核磁共振谱仪(Bruker,Mercury plus 400 MHz spectrometer)测试所得样品的1H-NMR;用VECTOR 22型红外光谱仪(Bruker AXS Co.,Ltd.,Germany)测试样品的红外光谱,KBr压片,波段为4000-400 cm-1;样品的质谱是在Bruker MALDI-TOF(基质辅助激光解吸电离飞行时间)质谱仪(Billerica,MA,USA)上进行的。
2 结果与讨论
2.1 核磁共振氢谱分析
反应原料PEG、SA和产品S1的核磁共振氢谱测试采用氘代DMSO-d6作溶剂,结果如图1所示。溶剂峰位于2.50 ppm处,0 ppm为内标TMS(四甲基硅烷)的化学位移。在图1(a)中,PEG的1H-NMR峰归属如下:a处,δ3.36-3.69 ppm(4H,m,-CH2-CH2-);b处,δ4.55 ppm(1H,t,-OH);在图1(b)中,c处为SA的化学位移(2.85 ppm);在图1(c)中,PEG-biCOOH的1H-NMR峰归属如下:e处δ3.43-4.15 ppm(nH,m,PEG中-CH2-CH2-);d处,δ2.41-2.58 ppm(nH,s,-CH2-CH2-);f处,δ12.1 ppm(1H,s,-COOH)。DMSO的溶剂残留峰与PEG-biCOOH的d处峰有所重叠,化学位移为12.1 ppm是羧基的特征峰。图1(c)中的d、f峰的出现表明开环的丁二酸酐已经接在PEG两端[21,25,26],样品经乙醚沉淀、二氯甲烷萃取及三次水透析后无其他杂质。
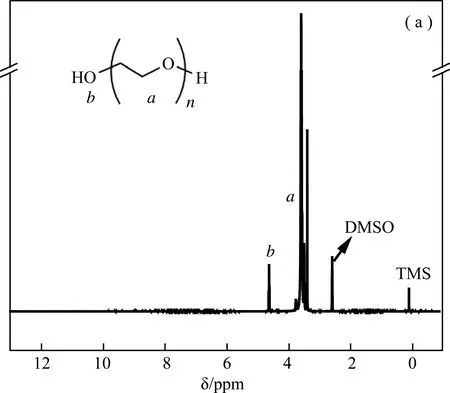
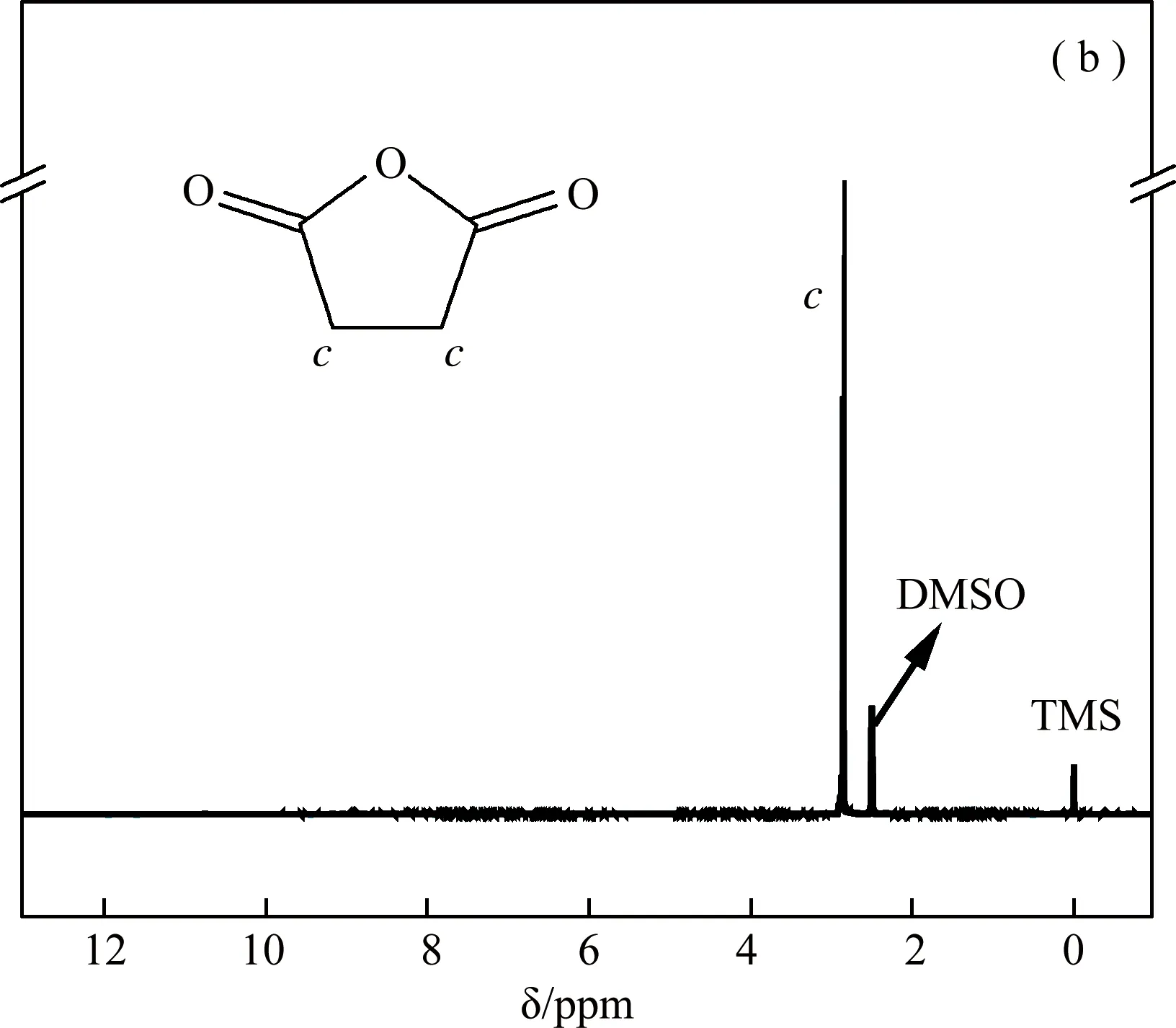

图1 (a)PEG、(b)SA和(c)PEG-biCOOH产品S1的核磁共振氢谱
2.2 红外光谱分析
PEG和S1的红外光谱如图2所示。其中,3430 cm-1处的宽吸收峰是由水中的羟基峰伸缩振动引起的。对于PEG,2888 cm-1处为-CH2-的对称和不对称伸缩振动峰;1467 cm-1处的中强吸收峰为-CH2-的变形振动峰;-CO-C-的伸缩振动峰在1109 cm-1处;1342、1280、962和842 cm-1处表示C-C骨架伸缩振动峰及-C-H-的变形振动峰。对于PEG-biCOOH,PEG本身的振动峰都与之相同,-CO的伸缩振动峰在1732 cm-1处[25,27],为酯的特征吸收峰,而PEG本身没有此峰,这证明了丁二酸酐与PEG端基羟基键合成功。
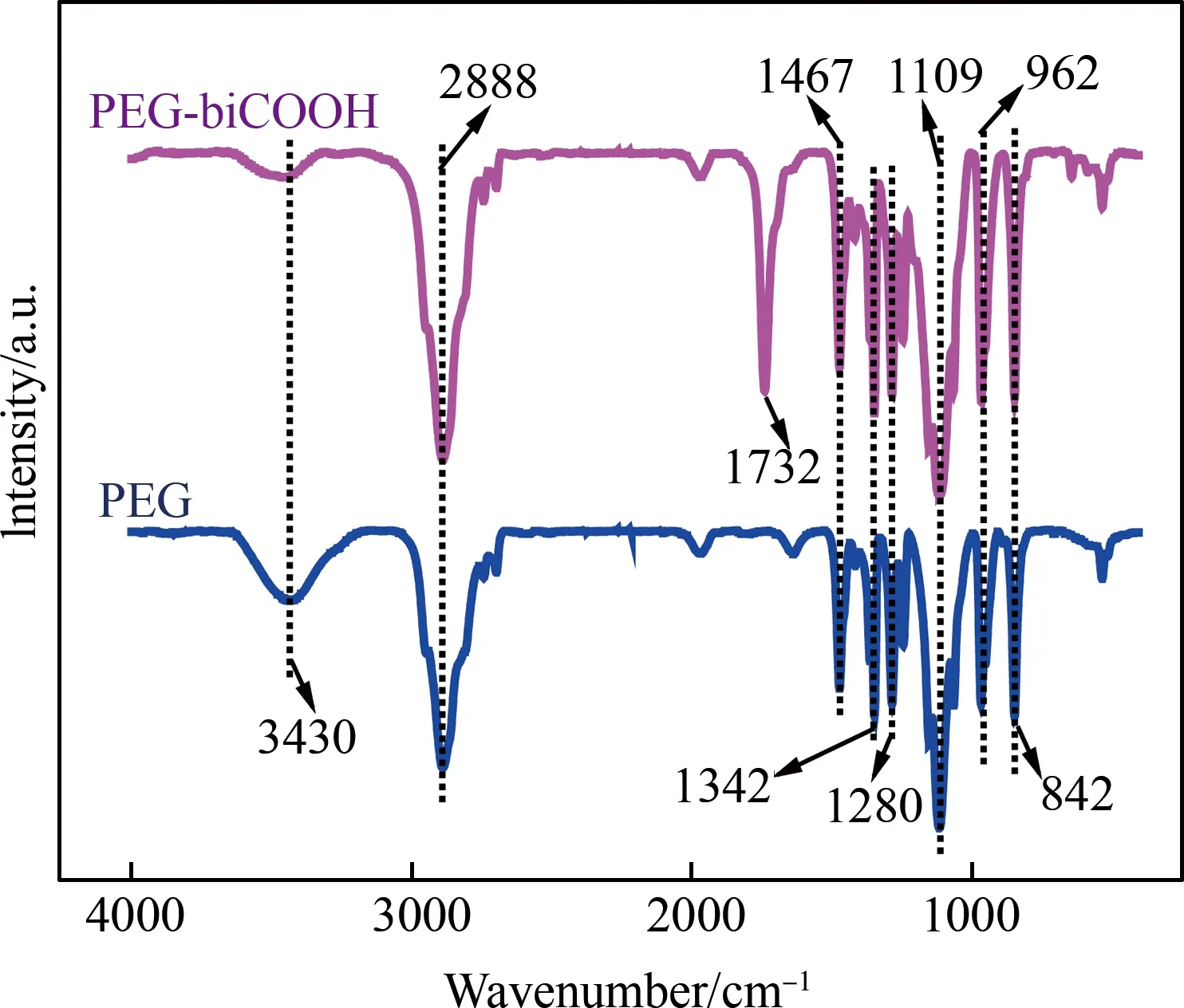
图2 PEG及产品S1的红外光谱图
2.3 MALDI-TOF质谱分析
采用MALDI-TOF质谱法测定了PEG和产品S1的分子量(图3)。如图所示,PEG和PEG-biCOOH的平均分子量分别为2000-2206 u。且PEG-biCOOH的分子量与理论值一致(~2200),表明丁二酸酐与聚乙二醇两端均被键合。
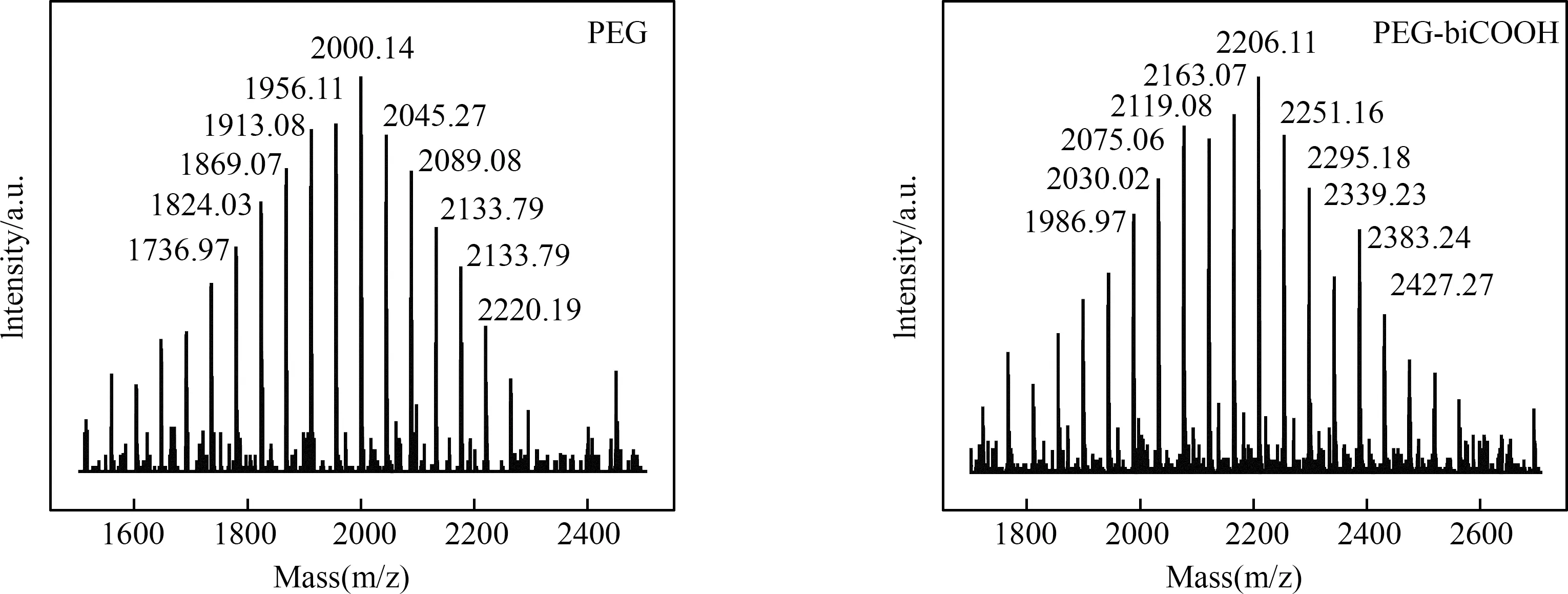
图3 PEG及产品S1的MALDI-TOF MS质谱图
2.4 不同溶剂对聚乙二醇与丁二酸酐反应的收率的影响
活化PEG常用的溶剂有DMF、甲苯和二氧六环[20,21,23]。为考察不同溶剂对PEG-biCOOH收率的影响,分别用二氧六环和甲苯替代DMF作为反应溶剂,其他条件与S1合成条件一致,编号为S2和S3。如表1所示,S1和S2透析前后的平均收率(97.5、86.7%和94.6、85.1%)相差不大,明显高于S3(87.8、80.6%)。另外,从溶剂毒性上考量,二氧六环对皮肤、眼部和呼吸系统均有刺激性,并且可能对肝、肾和神经系统造成损害,急性中毒时可能导致死亡;甲苯对皮肤、粘膜有刺激性,高浓度气体对中枢神经系统有麻醉作用。鉴于甲苯和二氧六环做溶剂危险性较大,而DMF无色、价格较低、沸点较高(153 ℃)不易挥发,毒性低,且收率高,故选择DMF为反应溶剂。
2.5 反应摩尔比对收率的影响
改变反应中加入的SA摩尔量,保持其他条件和S1一致,考察一系列PEG与SA的反应摩尔比对产品收率的影响。加入SA的量为4.4、4.8、5.2、6.0、6.4、6.8、7.2、7.6、8.0和8.4 mmol时,对应的产品编号分别为S4-S13,与S1(SA的摩尔量为5.6 mmol)作比较,结果如图4所示。随着加入的SA摩尔量的增加,透析后PEG-biCOOH的收率逐渐增加;当SA增加到5.6 mmol时(产品S1),PEG与SA的反应摩尔比为1:2.8,收率达到峰值,为86.7%;然后,随着SA的量的继续增加,收率逐渐下降至67.2%(产品S14)。各产品透析前收率列于表1,与图3趋势一致。
实验过程中发现,随着SA浓度的增加,反应物混合溶液的颜色也出现了相应的变化:S4-S10号反应溶液为浅黄色,S11-S13号反应溶液为深黄色,S14号反应溶液则开始变为酒红色,收率急剧下降。经纯化,所得样品均为白色絮状粉末,其核磁共振氢谱和红外光谱结果与最优条件下所合成样品一致。
2.6 反应时间对收率的影响
在PEG与SA的摩尔比为1:2.8,反应溶剂为DMF,反应温度为60 ℃的条件下,考察了不同反应时间对产品收率的影响。反应时间分别为3、6、9、12、18、36和48 h时,样品编号为S14-S20,与S1(反应时间24 h)作对比,结果如图5所示。随着反应时间的增加,透析后PEG-biCOOH的收率逐渐增加;当反应时间达到24 h时(产品S1),收率达到峰值,为86.7%;延长反应时间至36 h和48 h时,其产品收率基本不变,分别为~86.5(S19),~86.6%(S20)。各产品透析前收率列于表1,透析前PEG-biCOOH的收率与图4趋势一致,说明24 h即为此反应的最优反应时间。

表1 各产品的反应条件及其收率
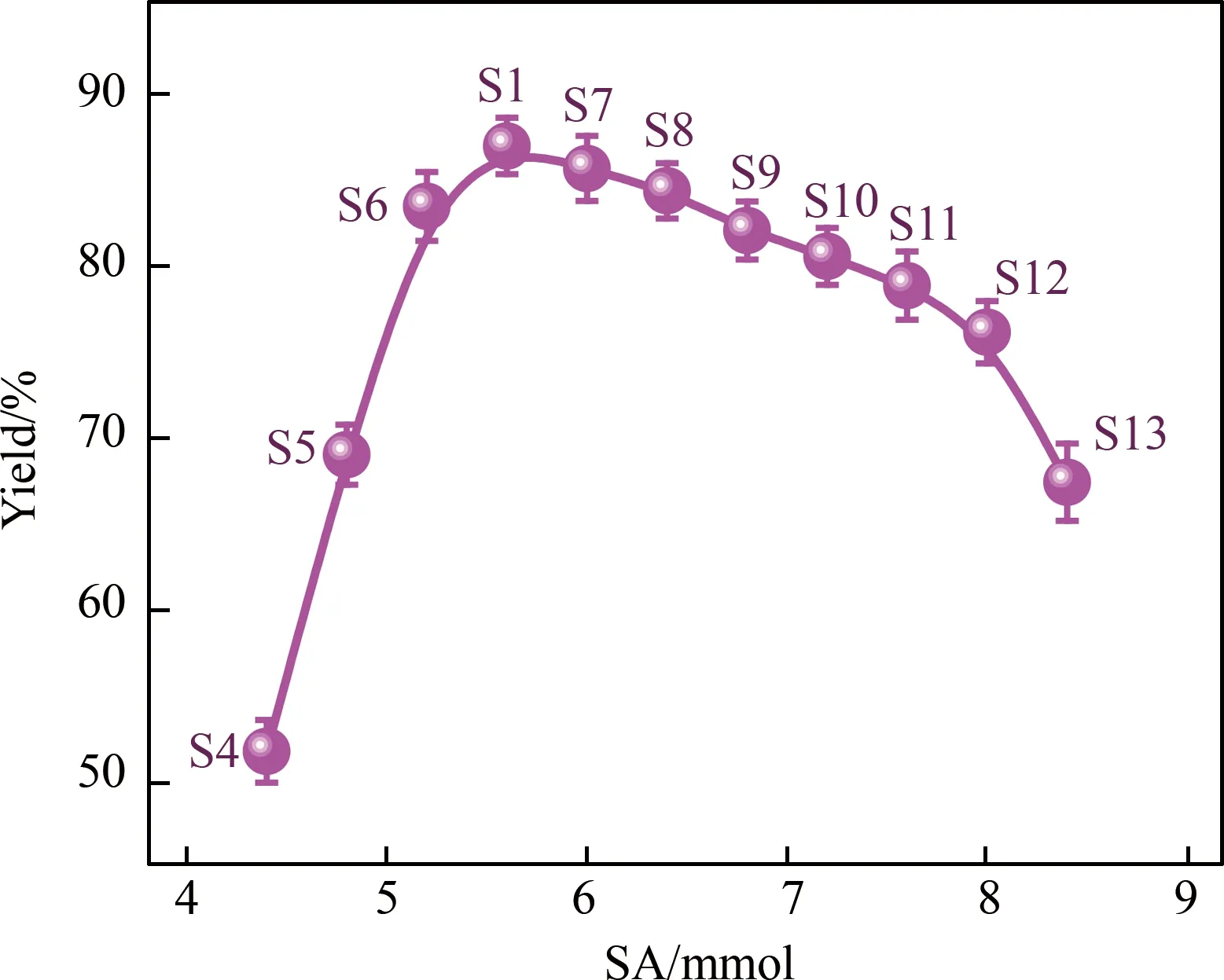
图4 SA的摩尔量对透析后收率的影响

图5 反应时间对透析后收率的影响
2.7 反应温度对收率的影响
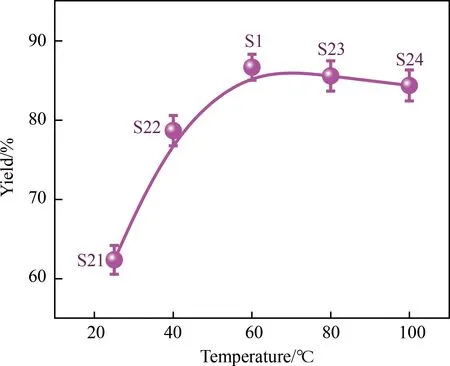
图6 反应温度对透析后收率的影响
反应溶剂为DMF,反应时间为24 h,反应摩尔比例PEG:SA为1:2.8时,考察了不同反应温度对产品收率的影响。反应温度分别为25、40、80和100 ℃,所得样品编号为S21-S24,与S1(反应温度60 ℃)做对比,结果如图6所示。从25 ℃升高至60 ℃,透析后平均收率分别为~62.4,~78.7,~86.7%,60 ℃时达到最大收率(产品S1)。反应温度为80和100 ℃时,其反应溶液的颜色由浅黄色变为黄色,制得的产品收率逐渐降低,分别为~85.6(S23),~84.4%(S23)。各产品透析前收率亦列于表1,均表明了60 ℃为最优反应温度。
3 结论
以4-二甲氨基吡啶为催化剂,采用聚乙二醇(PEG,平均分子量2000 u)和丁二酸酐(SA)反应制备了聚乙二醇二羧基醚(PEG-biCOOH)。考察了溶剂、投料配比、反应时间和反应温度对产品收率的影响,得出最优反应条件:反应溶剂为DMF,PEG与SA的摩尔比为1:2.8,反应温度是60 ℃,反应时间为24 h,此条件下的产物透析后的平均收率为86.7%。核磁共振氢谱、红外光谱和质谱测试结果均表明PEG端基羟基与开环后的丁二酸酐成功键合,制得PEG-biCOOH。相比于传统方法,该方法的优点是反应温度低,容易操作,溶剂毒性低,反应条件温和,收率较高。PEG-biCOOH的合成为进一步与抗癌药物反应合成前体药物、制备药物纳米颗粒等提供了一定的研究基础。
