酚羟基席夫碱钯配合物的合成、表征及其催化性能
2021-01-28郭丽娜闫丽岗高媛媛韩利民
郭丽娜, 闫丽岗, 高媛媛, 韩利民
(内蒙古工业大学 化工学院,内蒙古 呼和浩特 010051)
Suzuki-Miyaura反应是通过C—C键的形成构建联苯衍生物最有效的方法[1],通常在Pd催化剂的作用下,有机硼试剂与卤代芳烃或烯烃通过交叉偶联反应来完成。该反应被广泛应用于药物、天然产物、聚合物、有机电致发光材料等的合成中[2-5]。可见Pd催化剂的性能对该类反应的发生起着重要作用,其中配体的选择又是影响催化效率的关键因素[6]。因此,为了提高Pd催化剂的催化效率,研究者一直致力于寻找更有效的配体体系用于Suzuki-Miyaura反应。
Scheme 1
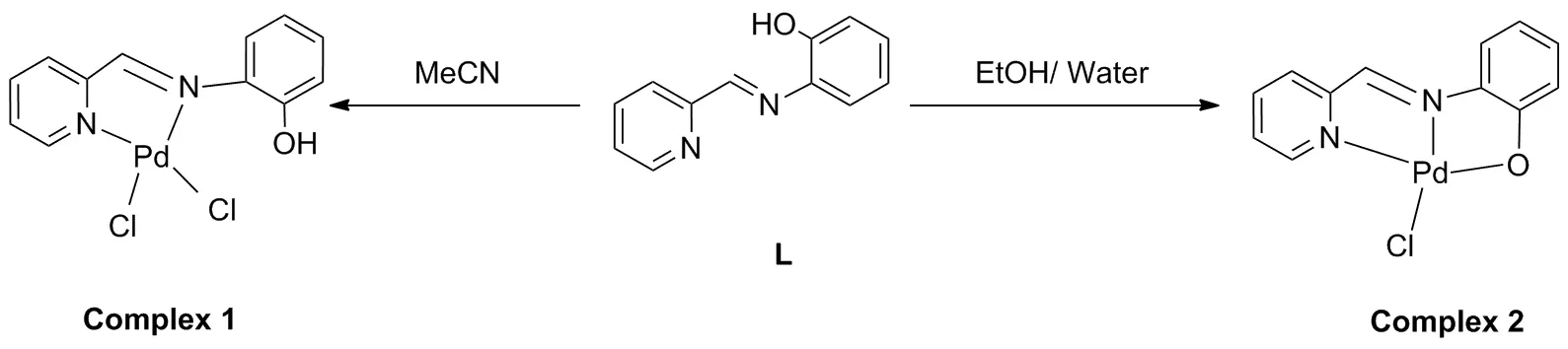
多年来,由于磷配体和卡宾配体的结构和电子性质的可调性而得到了广泛的应用,但由于它们在空气中的不稳定性且难以处理,仍不是钯催化的理想配体[7-8]。因此,开发新型的无磷或无碳的化合物并且能够在环境友好的条件进行Suzuki-Miyaura反应是十分必要的。其中含氮配体构建的Pd催化剂得到了大量的报道,例如,氮-杂环、席夫碱、肟、腙类衍生物[9-11]。这类催化剂因其价格低廉、无毒、在空气中稳定、活性好而被用于Suzuki偶联反应。值得注意的是,席夫碱作为一种重要的催化剂配体,不仅可以像磷配体一样灵活地调节电子和空间性质,而且合成简单,对空气不敏感。特别是席夫碱配体的配位方式受到与席夫碱结合的不同N杂环的调节,因此其催化作用也容易得到相应的改变[12-13]。同时,辅助配体对钯催化剂的调节也起着一定的作用,卤素是钯催化中使用的一类辅助配体,卤素的类型及数量对Suzuki-Miyaura反应会产生不同的催化效果。
众所周知,在钯催化剂催化Suzuki-Miyaura反应中,首先钯催化剂要从Pd(II)的稳定状态解离为单配体配位的Pd(0)活性中间体,进而Pd(0)活性中间体经过氧化加成、金属转移、还原消除3个历程完成催化,因此,钯催化剂的催化效率受到Pd(II)/Pd(0)解离效率的影响[14]。本课题组先前研究了具有不同供电子能力的吡啶辅助配体对钯催化剂催化效率的影响,结果表明,吡啶配体供电子能力的强弱会影响其与金属钯连接的键能,进而影响钯配合物的稳定性。当辅助配体的供电子能力较强时,其从钯催化剂中的解离效率就会降低,这会阻碍零价钯活性物质的生成,从而降低偶联反应的产率[15]。本文合成一个氨基酚类的三齿席夫碱配体,通过改变溶剂调控配位方式,得到两个含有不同氯离子数量的钯配合物,并通过配合物的单晶结构对不同配位模式下钯配合物的结构进行比较,并通过核磁数据分析了不同配位结构对钯中心电子云密度的影响[16]。
1 实验部分
1.1 仪器与试剂
XT-4型显微熔点仪; Agilent 500 MHz型核磁共振仪(DMSO-d6或CDCl3为溶剂, TMS为内标);Nicolet FT-IR 型红外光谱仪(KBr压片); Elementar var Ⅲ型元素分析仪; Shimadzu LCMS-2020型质谱仪;ruker SMART APEXⅡ 型X射线单晶衍射仪;Bruker D8 Venture X型射线单晶衍射仪。
所用试剂均为分析纯或化学纯。
1.2 配体及配合物的合成
(1) (2-NC5H4)C(H)=N(C6H4OH-2)(L)的合成
称取2-氨基苯酚110 mg(1.0 mmol)溶解于10 mL甲醇溶液中,加入吡啶-2-甲醛120 μL(1.2 mmol)于60 ℃搅拌1 h。反应结束后,减压蒸除溶剂,所得油状物中加入10 mL正已烷,静置于冰箱中过夜后过滤,干燥得淡黄色针状固体180 mg,收率90.9%, m.p.102~106 ℃;1H NMR(DMSO-d6, 500 MHz)δ: 9.22(s, 1H), 8.71(q,J1=2.0 Hz,J2=1.0 Hz, 1H), 8.70(q,J1=1.0 Hz,J2=1.0 Hz, 1H), 8.40(d,J=8.0 Hz, 1H), 7.95(td,J=7.5 Hz, 1H), 7.52(td,J=6.0 Hz, 1H), 7.30(dd,J=5.0 Hz, 1H), 7.13(td,J=7.0 Hz, 1H), 6.94(d,J=6.5 Hz, 1H), 6.86(td,J=7.5H z, 1H);13C NMR(DMSO-d6, 125 MHz,)δ: 159.78, 154.94, 151.84,149.90, 137.24, 128.68, 125.82, 121.92, 120.11, 120.01, 116.75; IRν: 3362,1627,1587,1152,739 cm-1; Anal. Calcd for C12H10N2O: C 72.71, H 5.08, N 14.13, found C 71.39, H 4.87, N 13.86; MS(ESI)m/z: Calcd for C12H10N2O{[M+H]+} 199.2, found 198.22。

Scheme 2
(2) [Pd{2-(NC5H4)C(H)=N[2-(OH)C6H4]}(Cl2)] (Complexe1)的合成
称取L19.8 mg(0.1 mmol)溶解于5 mL乙腈中,缓慢滴加含有Na2PdCl429.4 mg(0.1 mmol)的5 mL乙腈溶液,60 ℃搅拌36 h。反应结束后,过滤,滤液静置于室温下缓慢蒸发,大约一周后得粉红色块状固体29.6 mg,收率79%;1H NMR(DMSO-d6,500 MHz)δ: 10.05(s, 1H), 9.07(d,J=5.5 Hz, 1H), 8.73(s, 1H), 8.40(t,J=7.5 Hz, 1H), 8.22(d,J=7.5 Hz, 1H), 7.95(t,J=6.5 Hz, 1H), 7.17(t,J=9 Hz, 2H), 6.93(d,J=10.5 Hz, 1H), 6.84(t,J=7.5 Hz, 1H);13C NMR(DMSO-d6, 125 MHz)δ: 174.31, 156.01, 150.57, 149.51, 141.58, 135.35, 125.55, 116.56, 109.93;IRν: 3473.96,1599.17 cm-1;Anal. Calcd for C12H10N2OCl2Pd: C 38.38, H 2.68, N 7.46; found C 38.32, H 2.64, N 7.38; MS(ESI)m/z: Calcd for C12H10N2OCl2Pd{[M+Na]+}398.3, found 375.52。
(3) [Pd{2-(NC5H4)C(H)=N[2-(O)C6H4]}(Cl)](Complexe2)的合成
称取L 19.8 mg(0.1 mmol)溶解于5 mL乙醇中,缓慢滴加含有Na2PdCl429.4 mg(0.1 mmol)的5 mL水溶液, 60 ℃搅拌36 h。反应结束后冷却至室温,过滤得红色固体32.6 mg,收率88%;1H NMR(DMSO-d6,500 MHz)δ: 8.44(s, 1H), 8.35(d,J=5.5 Hz, 1H), 8.14(t,J=7.5 Hz, 1H), 7.76(d,J=8.0 Hz, 1H), 7.6(t,J=7.0 Hz, 1H), 7.38(d,J=7.0 Hz, 1H), 7.04(t,J=10.0 Hz, 1H), 6.47(m, 2H);13C NMR(125 MHz, DMSO-d6)δ: 176.42, 161.79, 152.15, 151.78, 142.10, 135.78, 134.76, 128.09, 120.41, 119.36, 116.20; IRν: 1574.24 cm-1;Anal. Calcd for C13H13N2O2ClPd: C 42.06, H 3.53, N 7.55; found: C 42.01, H 3.22, N 7.32; MS(ESI)m/z: Calcd for C12H9N2OClPd{[M+Na]+}360.3, found 337.92。
1.3 钯配合物催化活性
依次称取4-碘代甲苯218 mg(1 mmol)、苯硼酸133 mg(1 mmol)、K3PO442 mg(1mmol)、钯催化剂(0.5%)溶解于8 mL乙醇中,78 ℃反应4 h。反应完成后,减压蒸馏除去溶剂,经硅胶柱层析(洗脱剂:正已烷)纯化得交叉偶联产物4-甲基联苯: 淡黄色固体,m.p.49~52 ℃;1H NMR(CDCl3, 500 MHz)δ: 7.58(d, 2H), 7.48(d, 2H), 7.42(t, 2H), 7.32(t, 1H), 7.23(d, 2H), 2.39(s, 3H)。联苯: 白色固体,m.p.69~70 ℃;1H NMR(CDCl3, 500 MHz)δ: 7.59(m, 4H), 7.44(m, 4H), 7.33(m, 2H)。
1.4 配合物结构
Complexe1的单晶结构采用德国Bruker SMART APEXⅡ 型X射线单晶衍射仪,Complexe2的单晶结构采用德国Bruker D8 Venture X型射线单晶衍射仪,Mo-K/α为射线源(λ=0.71073 Å),SHELXS-97程序直接法解析结构,用SHELXL-97在F2进行全矩阵最小二乘优化[17-18]。
2 结果与讨论
2.1 钯配合物的合成与表征
配体及钯配合物的合成(Scheme 2),配体按照文献[19]方法合成,配体中的亚胺氮、吡啶氮和酚羟基氧均有较好的配位能力,因此该配体是一个性能优良的三齿配体,且配位形式比较灵活。将该配体与Na2PdCl4在相同的条件下反应,当溶剂为乙腈时,酚羟基的氧没有参与配位,形成了以钯原子为中心,4个配位原子分别为亚胺的氮、吡啶的氮和两个氯原子;而当以乙醇和水作为溶剂时,酚羟基发生了去质子化作用,此时的4个配位原子分别为亚胺的氮、吡啶的氮、酚羟基的氧和一个氯原子,可见加入不同的溶剂进行反应,得到结构不同的两个钯配合物。在这个过程中,溶剂起到了关键作用[20]。相关研究指出,酸碱作用会导致配位模式从κ2-N,N 到 κ3-N,N,O的转换[21]。按照这一理论,将Na2PdCl4溶解在不同的溶剂中,然后对其pH值进行测试。结果显示,当Na2PdCl4溶解于乙腈时,溶液的pH值为1.89,而当Na2PdCl4溶解与乙醇和水的混合溶液(1/1)时,pH值显示为3.7。这表明当乙腈作为溶剂时,体系整体显示偏酸性,当乙醇和水为溶剂时,体系比乙腈体系碱性强一些。

表1 配合物1和配合物2的晶体数据及结构参数
同时,该配体及配合物在空气中放置一段时间不会变质,有着较好的环境稳定性。对其进行了元素分析、红外和核磁的表征。对比两个配合物的核磁氢谱,在Complexe1中,δ10.05为酚羟基中氢原子的特征峰,而在Complexe2中,δ10.05附近没有出现吸收峰,说明酚羟基中氢原子的特征峰并未出现,这表明酚羟基的氧原子参与了配位,形成了Pd—O键。

图 1Complexe 1和Complexe 2 的分子结构

表2 催化剂在Suzuki-Miyaura反应中的催化评价
2.2 钯配合物的结构
Complexe1(CCDC: 2025866)和Complexe2(CCDC: 2025867)的分子结构如图1所示,钯配合物的晶体数据如表2所示。
通过对比两个钯配合物的单晶结构可知,两个配合物的共同点是均为四配位平面四边形结构,在Complexe1中,与钯配位的4个原子分别为Niminic、 Cl1、 Cl2和NPy,所形成的扭角为5.009(634)°;而在Cmplexe2中,与钯配位的4个原子分别为Niminic、 O1、 Cl1和NPy,所形成的扭角为0.566(73)°,因此 ,Complexe2中4个配位原子的平面化程度更高一些。相反地,两种配位模式导致的结构差异主要有两个方面:首先,在Cmplexe1中,与钯配位的4个原子所形成的键长分别为:2.020(2)(Pd1—Niminic)、 2.020(2)(Pd1—NPy)、 2.289(7)(Pd1—Cl1) 和 2.303(7)(Pd1—Cl2);在Complexe2中,与钯配位的4个原子所形成的键长分别为:1.944(2)(Pd1—Niminic)、 2.014(2)(Pd1—NPy)、 2.015(2)(Pd1—O1)和2.3004(11)(Pd—Cl1)。这些数据表明当Pd—O键取代Pd—Cl键时,Complexe2中的键长均有不同程度的缩短。其次,在Complexe1中,配位的五元螯合环(环1: Pd1—Niminic—C7—C8—NPy)与苯环所成的二面角为59.766(749)°,当Pd—O键形成后,又形成了一个新的五元环(环2: Pd1—Niminic—C7—C12—O1),同时由于配位作用,苯环发生了一定程度的转动,这使得环1与苯环之间的二面角变为1.778(85)°。环1、环2、吡啶环、苯环4个环中,吡啶环和苯环之间的二面角最大,为4.069(100),也就是说,氧原子的配位使得Complexe2中所形成的4个环基本处于同一个平面[22-23]。
2.3 催化反应研究
两个钯配合物的催化性能通过Suzuki-Miyaura反应进行评价。使用苯硼酸和4-碘代甲苯作为反应模型,乙醇作为溶剂,加入适当的K3PO4作为碱,反应在空气状态下回流4 h。表2列出了反应条件及催化产物4-甲基联苯和联苯的GC产率。
由表2中数据可知,两个钯催化剂的催化产率分别为98.72%和92.31% ,催化效果较好,Complexe1的催化效率略高于Complexe2,这表明随着配合物中辅助配体氯离子个数的减少,催化剂的催化能力会减弱。按照钯催化剂催化Suzuki-Miyaura反应机理可知,催化剂催化能力的强弱取决于零价钯的产生效率,零价钯的产生效率和催化剂的稳定性有关[24]。课题组前期对含有不同副配体的化合物进行研究也证实,化合物越稳定,其产生零价钯的能力越弱,催化效率就会降低。相关研究表明[25],对于该类型的席夫碱配体,形成N—N—O配位模式的化合物比N—N配位模式的化合物会使得结构更为稳定,也就是说一个氯的配合物的稳定性高于两个氯的配合物,这一点也可由上述单晶数据证实,一个氯的钯配合物中与钯连接的4个配位原子的键长更短一些,结构更稳固,不容易解离产生零价钯的活性中间体,相反,两个氯的钯配合物中与钯连接的4个配位原子的键长较长,两个氯的钯配合物稳定性相对较低,零价钯的产生效率增加,因此催化产率相应的提高。
同时,从表中可以看出,该类配合物在进行Suzuki-Miyaura催化反应时[26],反应的副产物分别为1.15%和1.08%,说明该类催化剂对此反应的选择性较高。另外,本文所进行的催化反应是采用乙醇作为溶剂在空气中进行的,这也说明该类配体的钯催化剂是一种绿色环保、温和高效的催化体系。
用相同的反应物在不同的溶剂中合成了两种不同的席夫碱Pd配合物,配合物的核磁共振和X射线衍射分析揭示了两种配位模式下的电子云密度分布和结构特征。对溶剂的研究显示,配位方式的差异源于反应体系不同的pH值。进一步,在空气气氛下,通过Suzuki-Miyaura反应考察了两种钯配合物的催化活性。高效的转化率说明该类钯催化剂是一种绿色、环保、温和的催化体系;同时,催化结果的差异表明,氯离子的数量对催化剂的活化有轻微的影响,连接到Pd中心的氯离子数量的增加可以降低配合物的稳定性,从而促进Pd(0)活性物质的生成,带来更高的催化效率。
