益津降糖颗粒质量标准改进研究
2021-01-25满小溪许春燕高光男杨德智闵俊哲
满小溪,许春燕*,高光男,杨德智,闵俊哲*
(1.延边大学 药学院, 吉林 延吉 133002; 2.延边朝药药业有限公司, 吉林 龙井 133400 )
益津降糖颗粒由吉林延边朝药药业有限公司生产,该颗粒由人参、白术(炒)、茯苓、仙人掌、甘草5味药材制成,具有健脾益气、生津止渴的功效,常用于治疗II型糖尿病.益津降糖颗粒于2002年正式获得新药证书, 2005年其质量标准由新药试行标准转为正式标准.在该颗粒的原有质量标准中,甘草、仙人掌在定性鉴别时存在斑点不清晰和无相应对照品斑点的问题,而且人参皂苷Re未采用2020年版《中国药典》[1]规定的方法进行测定(采用的是薄层斑点扫描法).为此,本文对益津降糖颗粒方剂中的甘草、仙人掌的定性鉴别进行优化,并对人参皂苷Re采用高效液相色谱(HPLC)进行含量测定改进,以此完善益津降糖颗粒的质量标准.
1 材料
1.1 仪器
高效液相色谱仪(Primaide)和紫外检测器,HITACHI公司;紫外灯分析仪(ZF-1),上海金鹏分析仪器有限公司;多功能超纯水系统(Unique-R20),厦门锐思捷水纯化技术有限公司;超声波清洗器(KQ -250DE),昆山市超声仪器有限公司;电子天平(AL-104),上海梅特勒-托利多仪器有限公司;电子天平(DV215CD),上海奥豪斯国际贸易有限公司;漩涡混合器(XH-C),金坛市白塔新宝仪器厂;电热恒温鼓风干燥箱(OHG -907385-Ⅲ),上海新苗医疗器械有限公司.
1.2 试剂
人参皂苷Re(批号为110754-201827)、人参皂苷Rg1(批号为110703-201933)、人参(批号为120917-201712)、甘草(批号为120904-201620)、仙人掌(批号为121283-201303),均购于中国药品生物制品检定所;益津降糖颗粒(批号分别为T20180801、T20180802、T20180901、T20180902), 由延边朝药药业有限公司提供;不含甘草的益津降糖颗粒阴性对照药品、不含仙人掌的益津降糖颗粒阴性对照药品、不含人参的益津降糖颗粒阴性对照药品,均由延边朝药药业有限公司实验室配制;硅胶G薄层板,青岛海洋化工厂分厂;聚酰胺薄膜板,浙江省台州市路桥四甲生化塑料厂;乙腈(色谱纯)、甲醇(色谱纯),天津赛孚瑞科技有限公司;无水乙醇,天津市科密欧化学试剂有限公司;超纯水,延边大学药学院实验室制备;三氯甲烷(分析纯),北京化工厂;正丁醇(分析纯)、丙酮(分析纯)、甲苯,沈阳市华东试剂厂;工业乙酸乙酯,延吉浩然化工有限公司;甲酸、氢氧化钠,上海阿拉丁生化科技股份有限公司.
2 检验方法
2.1 定性鉴别
2.1.1人参的定性鉴别
取10.058 6 g研细的装量差异项下的益津降糖颗粒内容物置于索氏提取器中,加入适量的甲醇后加热回流6 h;蒸干提取液,残渣加水30 mL,用水饱和正丁醇振摇提取5×20 mL;合并正丁醇提取液,用质量分数为2%的氢氧化钠溶液洗涤4×40 mL;去除氢氧化钠洗液后用50 mL正丁醇饱和水洗涤1次,去除水层后取正丁醇液;旋干正丁醇液,残渣加甲醇后将其转移至5 mL容量瓶中,稀释至刻度后作为供试品溶液.
取人参对照药材0.603 2 g,按照供试品溶液的制备方法制成对照药材溶液.另取对照品人参皂苷Rg1和Re,加甲醇制成1 mg/mL的混合溶液(作为对照品溶液).取不含人参的阴性对照品10.007 4 g,按照供试品溶液的制备方法制成阴性对照溶液.按照2020年版《中国药典》四部通则0502中的TLC方法对人参进行定性鉴别.吸取上述4种溶液各4 μL,分别点于硅胶G薄层板上,然后以三氯甲烷-乙酸乙酯-甲醇-水(体积比为15∶40∶22∶10)的下层溶液为展开剂(温度低于10 ℃)展开,晾干后喷硫酸-乙醇溶液(体积比为1∶9)显色,并在105 ℃下加热约3 min.供试品与对照药材、对照品的TLC结果如图1所示.
2.1.2甘草的定性鉴别
取10.027 6 g研细的装量差异项下的益津降糖颗粒内容物置于索氏提取器中,加入适量的乙醚后加热回流1 h;过滤,弃去乙醚液,药渣再加入适量的甲醇后加热回流3 h;过滤,蒸干提取液,残渣加40 mL水使其溶解;用水饱和的正丁醇振摇提取3×20 mL,然后合并正丁醇提取液;水洗3次,弃去水层,蒸干正丁醇液,残渣加甲醇使其溶解;将溶液转移至5 mL容量瓶中,定容、摇匀后将其作为供试品溶液.
取甘草对照药材1.007 2 g,按照供试品溶液的制备方法制成对照药材溶液.取不含甘草的阴性对照品10.026 6 g,按照供试品溶液的制备方法制成阴性对照溶液.按照2020年版《中国药典》四部通则0502中的TLC方法对甘草进行定性鉴别.吸取上述3种溶液各4 μL,分别点于硅胶G薄层板上,以甲苯-三氯甲烷-甲醇(体积比为5∶5∶1)为展开剂展开,晾干后喷硫酸-乙醇溶液(体积比为1∶9)显色,并在105 ℃下加热3 min.供试品与对照药材的TLC结果如图1所示.
2.1.3仙人掌的定性鉴别
取2.009 6 g研细的装量差异项下的益津降糖颗粒内容物置于索氏提取器中,加入体积分数为70%的乙醇并浸泡12 h;过滤,旋干滤液,然后加10 mL水使其溶解;用乙酸乙酯提取3×20 mL,然后合并乙酸乙酯提取液;过滤,旋干滤液,加入1 mL甲醇即得供试品溶液.
取仙人掌对照药材4.006 1 g置于索氏提取器中,加入适量的水后加热回流4 h;过滤,用乙酸乙酯提取3×20 mL,然后合并乙酸乙酯提取液;过滤,旋干滤液,加入1 mL甲醇即得对照药材溶液.
取不含仙人掌的阴性对照品2.006 7 g,按照供试品溶液的制备方法制成阴性对照溶液.按照2020年版《中国药典》四部通则0502中的TLC方法对仙人掌进行定性鉴别.吸取上述3种溶液各4 μL,分别点于同一聚酰胺薄膜板上,然后以丙酮-甲醇-水-甲酸(体积比为3∶3∶5∶1.5)为展开剂展开,晾干后喷三氯化铝-乙醇溶液(1 g三氯化铝溶于100 mL乙醇溶液)显色,并在105 ℃下加热3 min.供试品与对照药材的TLC结果如图1所示.
2.2 人参皂苷Re的含量测定
2.2.1对照品、供试品及阴性对照溶液的制备
1)对照品溶液的制备.精密称取人参皂苷Rg1对照品和人参皂苷Re对照品,然后加入甲醇制成0.2 mg/mL的混合溶液,摇匀后即得对照品溶液.
2)供试品溶液的制备.取2.002 3 g研细的装量差异项下的内容物置于索氏提取器中,加入适量的三氯甲烷后加热回流3 h;挥干溶剂后的药渣用50 mL正丁醇溶解,然后移入100 mL锥形瓶中;超声30 min,过滤,量取续滤液25 mL,蒸干;残渣加甲醇溶解,并转移至5 mL容量瓶中定容,摇匀,过滤,所得滤液即为供试品溶液.
3)阴性对照溶液的制备.称取不含人参的阴性对照颗粒2.008 0 g,与供试品同法制备即得阴性对照溶液.
2.2.2HPLC谱图的获取
色谱柱为Sepax Bio -C18 (4.6 mm×250 mm, 5.0 μm),检测波长为203 nm,柱温为40 ℃,流速为1.0 mL/min,进样量为5 μL.参考2020年版《中国药典》(一部)人参项下的条目及文献[2-9]对A、B两种流动相考察.A相为乙腈-水溶液,洗脱梯度为:0~35 min,19%乙腈;35~55 min,19%~29%乙腈; 55~70 min,29%乙腈;70~100 min,29%~40%乙腈.B相为乙腈-0.05%磷酸溶液(体积比为20∶80).人参皂苷Rg1、Re的标准品和供试品的HPLC图如图2所示.
2.2.3方法学考察
1)线性关系考察.分别取5 μL不同浓度的人参皂苷Re(0.01、0.05、0.1、0.2、1.0、2.0 mg/mL),然后按2.2.2色谱条件测定其峰面积.以峰面积的积分值为纵坐标、人参皂苷Re的浓度为横坐标绘制标准曲线,其线性结果如表1所示.
2)精密度试验.精密吸取5 μL进样(2.2.1中制备的人参皂苷Re对照品溶液),试验结果如表2所示.由表2可知,人参皂苷Re对照品溶液的RSD值为2.48%,这表明色谱系统的响应值具有良好的重复性.

表1 人参皂苷Re的标准曲线方程及检测限
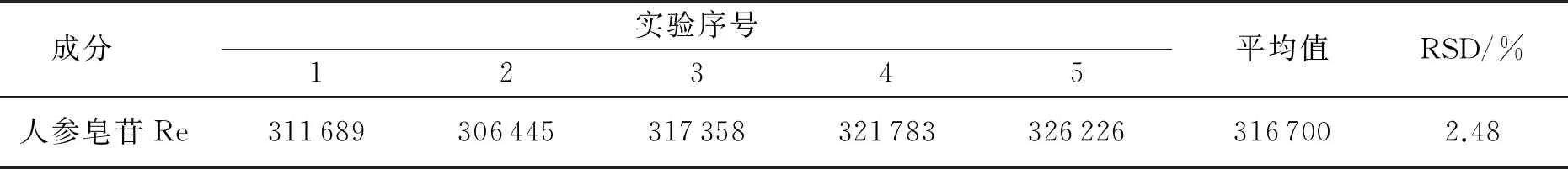
表2 人参皂苷Re精密度的试验结果 (n=5)
3)稳定性试验.精密吸取5 μL进样(2.2.1中制备的人参皂苷Re对照品溶液),并间隔2 h进样,结果如表3所示.由表3可知,在72 h内人参皂苷Re的峰面积的平均值为229 737, RSD值为1.22%,这表明人参皂苷Re在72 h内稳定.
4)专属性试验.分别吸取5 μL 2.2.1中制备的对照品溶液、供试品溶液和阴性对照溶液,按2.2.2中的色谱条件测定其HPLC, 结果如图3所示.由图3可以看出:供试品色谱中有与对照品一致的色谱峰,并且该峰与其他峰相分离;阴性对照溶液(空白试验)中未出现干扰峰,即不干扰主成分的测定.由此表明,上述实验的分析方法具有专属性.
5)加标回收试验.精密称取5 mg人参皂苷Re对照品,再称取研细的装量差异项下的内容物约2 g,然后将二者置于同一索氏提取器中;加入50 mL的三氯甲烷后加热回流3 h,然后将挥干溶剂的药渣连同滤纸移入100 mL锥形瓶中,加50 mL正丁醇,浸泡过夜;超声30 min(功率为250 W,频率为50 kHz),过滤;弃去初滤液,量取续滤液25 mL,旋干;残渣加入甲醇使其溶解后将其转移至5 mL容量瓶中,定容至刻度;摇匀,过滤,取续滤液按2.2.2中的方法进样,结果如表4所示.由表4可以看出,供试品溶液中的人参皂苷Re的回收率为85.40%~116.09%,平均回收率为92.88%, RSD值为12.43%.

表3 人参皂苷Re对照品溶液的稳定性试验结果 (n=5)
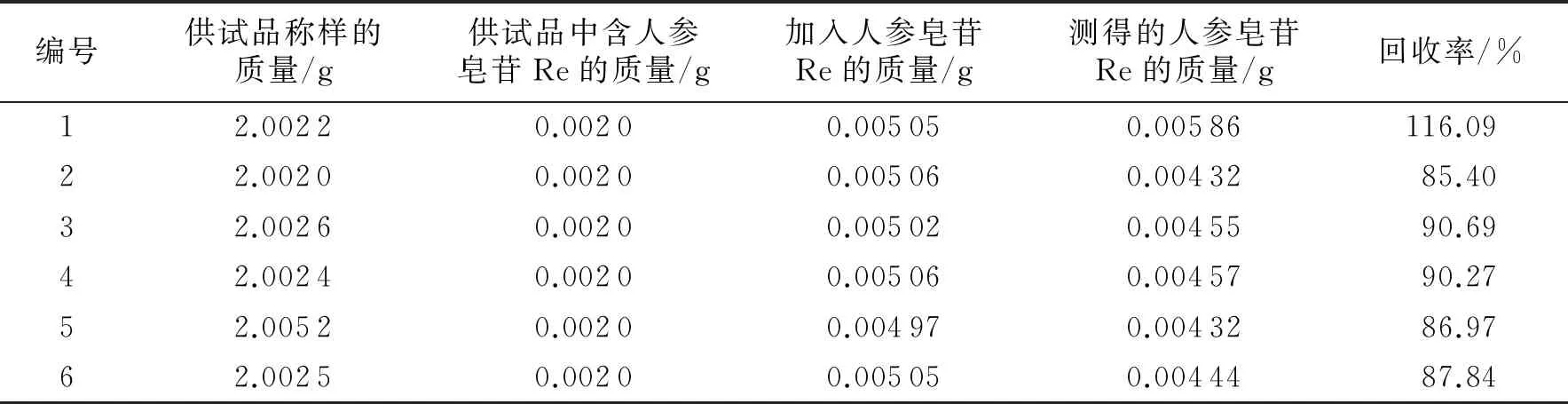
表4 益津降糖颗粒供试品的回收试验结果
2.2.4实际样品含量测定
按照2.2.2中的方法对4个批次的供试品(T20180801、T20180802、T20180901、T20180902)进行测定,结果如表5所示.由表5可以看出,人参皂苷Re的RSD值为2.88%,且每个批次样品中的人参皂苷Re的含量均大于1.2 mg(每袋益津降糖颗粒为5 g,且规定每袋中人参皂苷Re的含量不低于3 mg,因此2 g益津降糖颗粒应含人参皂苷Re不低于1.2 mg),由此表明益津降糖颗粒制剂中的人参皂苷Re的含量符合益津降糖颗粒的质量要求.

表5 供试品中人参皂苷Re含量的测定结果(n=3)
3 结果与分析
3.1 定性鉴别
本文选用2020年版《中国药典》规定的方法对甘草进行前处理.由该方法得到的甘草斑点纯净,通过斑点的深浅即可判定甘草的量.对3种展开剂(乙酸乙酯-甲酸-乙酸-水(体积比为15∶1∶1∶2)、甲苯-三氯甲烷-甲醇(体积比为5∶5∶1)和丙酮-水-甲酸(体积比为1∶1∶0.1))进行比较可知,当使用甲苯-三氯甲烷-甲醇为展开剂(体积比为5∶5∶1)时,供试品色谱在对照药材色谱的相应位置上显示出8个颜色相同的斑点,且斑点的分离度和显色均优于其他两种展开剂,同时阴性对照无明显干扰(见图1);因此,本文选择甲苯-三氯甲烷-甲醇(体积比为5∶5∶1)为展开剂.
采用体积分数为70%的乙醇溶液制备仙人掌对照药材溶液进行仙人掌定性鉴别时,供试品溶液、对照品溶液的斑点不在同一位置,不具有对应性;而采用水溶液制备仙人掌对照药材溶液进行仙人掌定性鉴别时,供试品溶液与对照品溶液的斑点出现在同一位置,即具有良好的对应性,这可能是70%乙醇的仙人掌提取物的溶解度优于水提取物的缘故[11-12],进而使仙人掌对照药材溶液中所含提取物的质量浓度高于供试品中所含提取物的质量浓度.在文献[13-15]研究基础上,本文利用不同展开剂(三氯甲烷-甲醇-甲酸(体积比为2∶3∶1)和丙酮-甲醇-水-甲酸(体积比为3∶3∶4∶1.5)对仙人掌的展开效果和不同比例甲酸对仙人掌展开斑点拖尾现象的影响进行了考察.考察结果显示,使用展开剂丙酮-甲醇-水-甲酸(体积分数比为3∶3∶4∶1.5)时,供试品色谱在对照药材色谱的相应位置上显示出6个相同颜色的规则斑点,且斑点的分离度和显色均优于另一种展开剂,同时阴性对照无明显干扰(见图1);因此,本文选择丙酮-甲醇-水-甲酸(体积分数比为3∶3∶4∶1.5)为展开剂.
3.2 定量测定
由图2可知,2020年版《中国药典》中规定的流动相和乙腈-0.05%磷酸水(体积比为20∶80)流动相均可以使人参皂苷Rg1、Re达到完全分离(分离度≥1.5).但是,使用2020年版《中国药典》中规定的流动相所获得的标准品和供试品的色谱图中Re的峰面积均显著大于Rg1的峰面积,且分析时间较长(人参皂苷Rg1、Re的出峰时间为45 min,梯度洗脱时间为115 min);而使用乙腈-0.05%磷酸水(体积比为20∶80)流动相,不仅可以明显改善Rg1和Re的吸收强度,使二者的峰面积接近,而且可减少分析时间(人参皂苷Rg1、Re的出峰时间为35 min,梯度洗脱时间为45 min).
4 结论
本文利用TLC方法,以甲苯-三氯甲烷-甲醇(体积分数比为5∶5∶1)和丙酮-甲醇-水-甲酸(体积分数比为3∶3∶4∶1.5)为展开剂,分别对益津降糖颗粒方剂中的甘草、仙人掌的定性鉴别进行优化表明,优化后的甘草和仙人掌在定性鉴别时其分离度、重现性、专属性均良好.利用HPLC方法(以Sepax Bio -C18 为色谱柱, 0.05%磷酸溶液-乙腈为流动相)对人参皂苷Re含量进行测定表明,该方法精密度、重复性、稳定性实验的RSD值均小于2.48%,同时阴性对照无明显干扰.另外HPLC方法的测定时间比2020年版《中国药典》中规定的方法缩短了70 min,且明显改善了2020年版《中国药典》规定方法中存在的人参皂苷Re和Rg1的色谱峰响应值差异较大的问题.综上,本文研究结果可为药企提高产品质量和检测速度提供参考.
