基于细菌16S rRNA 基因扩增子测序数据的系统发生树图制作方法
2021-01-21阎星羽廉振颖成丽娟刁玉涛王家林
丁 可,阎星羽,廉振颖,成丽娟,刁玉涛,王家林
(1.山东第一医科大学,山东 泰安 271016;2.山东第一医科大学基础医学院/基础医学研究所,山东 济南 250062;3.山东中医药大学第二附属医院,山东 济南 250001;4.山东第一医科大学附属肿瘤医院,山东 济南 250117)
细菌微生物16S rRNA 基因扩增子二代测序数据分析的后续任务之一是通过系统树形图(dendrogram/phylogenetic tree)展示微生物种群之间在进化或种系发生上的相互关系情况,dendrogram/phylogenetic tree 图分3 种类型,分别为进化分支图(cladogram),仅有拓扑结构,不能从数量上说明各分支间进化距离的大小;系统发生(发育)图(phylogram),各分枝长度表示碱基替换数,因而能从数量上说明各分支间进化距离的大小;时序图(chronogram)则用各分枝长度表示进化时间,其中系统发生图(phylogram)无疑是微生物分子遗传学研究最常用到的图形化方法[1]。目前系统发生图制作软件生成的系统发生树文件为Newick 和Nexus 格式,并且大多数系统发生树查看软件也主要兼容上述2 种文件格式,但Newick 和Nexus 格式的树文件只包含物种间的遗传距离和进化拓扑关系,结构比较单一。而R语言中的“ggtree”包对ape、ggplot2 等绘图包的功能进行了优化,“ggtree”除了能支持众多的树文件格式,其绘图方法更丰富,绘制的图形更准确和美观,最大的优点是能通过各种附加信息对树形图进行多方面的注释[2]。因此,本研究构建了微生物基因组扩增子二代测序数据的系统发生树形图的制作流程,通过QIIME 命令代码对测序数据进行预处理,聚类生成OTUs 表数据,对OTUs 表格数据进行筛选和转换,并用QIIME 分析管道(pipeline)的相关命令生成Newick 格式的系统发生树文件,以此文件为输入,用R 语言“ggtree”包的函数和代码绘制包含更多研究信息的不同类型的系统发生树图,现将结果报道如下。
1 材料与方法
1.1 数据来源 分析数据来自文献所用的原始测序数据[3],基于人体微生物在不同部位并随时间推移的变化,通过QIIME 分析方法,只选取其中的部分数据,即每天分别从2 个人的舌部、左手手掌、右手手掌和肠道共取得34 个标本,在Illumina HiSeq 2000 平台上进行微生物16s rDNA 扩增子测序。此外,从“https://data.qiime2.org/2017.6/tutorials/moving-pictures/emp -single -end -sequences/barcodes.fastq.gz”网站获得经过整理后的单向测序的数据。
1.2 软件系统 通过QIIME 完成系统发育树构建和可视化的工作流程,并使用是R 语言中的“ggtree”包(3.6.0 版)进行树文件的可视化和注释。
1.3 分析流程 构建可视化系统发生树的工作流程,关键步骤是形成BIOM(Biological Observation Matrix)格式的OTUs 表。QIIME 系统中有多个Python脚本用于生成OTUs 表,按聚类算法分为使用从头聚类方法(de novo)和使用参考数据库聚类方法。在此工作流程中,本研究使用的是基于Greengene 参考数据库的封闭参考(Closed reference)聚类分析方法生成OTUs 表数据进行分析的,具体步骤如下:①生成OTUs 表文件(otu_table.biom):目的是依据上一步生成的序列文件seqs.fna,通过与最新的Green-Genes 数据库比对进行聚类分析,其主要输出是OTUs 表格(out_table.biom),表格中的内容为记录每个OTU 在每个样品(微生物群落标本)中被观察到的次数;②OTUs 的过滤:通过filter_otus_from_otu_table.py 命令代码筛选OTUs,只保留相对含量>1‰(或其它比例)的OTUs;③OTUs 表文件格式转换:目的是将上一步生成的biom 格式的OTUs 表文件转换为纯文本(csv、tsv 或txt 格式)文件,利于下一步数据处理;④选取代表序列:由于每个OTU 中的序列不完全相同,因此需要通过pick_rep_set.py 脚本选取一条代表性序列作为该OTU 的序列,用于后续分析;⑤物种分级注释:用assign_taxonomy.py 脚本命令对每个OTU 代表性序列进行物种分类信息的注释,可以认为每个OTU 近似为一个物种,反之一个分类到“种(species)”或“属(genus)”水平的物种类别可以对应一个到多个OTU;⑥代表序列的比对:QIIME 系统的align_seqs.py 脚本提供3 种序列比对方法,即Py-NAST、MUSCLE 和INFERNAL,本研究中使用QIIME 系统默认的PyNAST 方法,它基于NAST 算法,将输入序列与提供的参考序列数据比对,在数据库中找到最高匹配的序列;且MUSCLE 不需要提供参考序列,可用于真菌转录间隔区(internal transcribed spacer,ITS)测序比对分析;而INFERNAL 利用RNA结构和序列相似性进行比对,与PyNAST 一样需要比对数据库;⑦筛选比对序列:由于上述align_seqs.py 脚本通过将长度200~400 bp 的目的序列和16S rRNA 基因的全序列比对,因此,生成的代表性序列包含空缺(gaps),为了保留代表性序列中的有用信息以构建系统发育树,需要通过filter_alignment.py 脚本对上述代表性序列进行筛选,去除碱基空缺等无用信息;⑧建树:运用python make_phylogeny.py 脚本构建系统发育树;⑨树文件的图形化:通过FigTree软件和R 语言“ggtree”包对Newick 格式的系统发育树文件进行图形化处理。系统发育树文件生成与可视化的数据处理工作流程见图1。
2 结果
本结果来自对34 个取样标本16S rRNA 基因测序数据的分析,34 个标本分别来自人体的不同部位和不同取样时间,为方便起见,本分析流程没有考虑采样时间,仅将不同取样部位作为分组因素进行分析。首选通过QIIME-1.9.1 系统的pick_closed_reference_otus.py 脚本生成OTUs 表和系统发生树文件,然后根据物种分类信息,将物种名称标记到“树叶”上,构建优化的物种聚类树文件,该树文件作为“ggtree”包的输入文件,再结合研究设计的分组信息绘制不同类型的系统发生树图。
2.1 样本信息和OTUs 聚类结果 本示例数据来自人体的肠道(gut)、左侧手掌(left palm)、右侧手掌(right palm)、和舌部(tongue),为了分析方便,省略了取样时间、抗生素使用情况等其它分组信息。通过封闭参考(closed reference)聚类法将有效测序序列按照≥97%的相似性归为一个OTU。采用RDP 算法与Greengene 16S rRNA 数据库比对,并将各OTU注释到所属的分类单元。使用Greengene 数据库可以将大多数OTU 序列注释到“属”水平,少部分OTU可以鉴定到“种”水平。通过上述方法将示例数据中的共177882 条序列聚类成4403 个OTU,在“门”和“属”两个水平上对同类的OTU 进行分类鉴定,分别归属于24 个门,615 个属,有关样本的人体部位分组情况、OTU 聚类和种属鉴定的具体情况见表1。OTUs 聚类及分类鉴定结果与同时生成的系统发生树文件,可以作为后续统计分析,构建系统发生树图的基础。

图1 系统发育树文件生成与可视化的数据处理工作流程

表1 样本测序序列与OTU 聚类情况统计表
2.2 各OTU 的物种归类 人体不同部位的微生物构成存在显著差异,通过QIIME 系统的种群多样性分析命令集core_diversity_analyses.py 可全面分析不同取样部位之间物种的两类多样性指数Alpha 多样性指数(α-diversity)与Beta 多样性指数(β-diversity)的差异,更重要的是可以发现人体特定部位的菌群相对含量(丰度)并发现其优势菌群。在“门”和“属”水平对菌群分类结果显示,具体菌群分类与丰度信息可以在多样性分析输出目录(“taxa_summary_plots”)中查看,该命令集还会给出不同物种的丰度在样本组(人体部位)之间差异的统计学假设检验结果(“group_significance_SampleType.txt”),其中拟杆菌属(bacteroides) 在肠道中的相对含量为60.48%,远高于其它身体部位(P<0.05)而链球菌属(streptococcus)只存在与手掌部和舌部,在肠道未检出(P<0.05),见图2。
2.3 QIIME 系统生成的原始系统发生树 FigTree 软件打开原始系统生成树文件,FigTree 的运行需要JAVA 语言支持,需根据软件提示安装相应的JAVA运行环境,依次点击file--open--rep_set3.tre 输入原始树文件rep_set3.tre,运行后生成rectangular、polar 和radial 3 种类型的系统发生树图,结果显示其只有物种间的进化距离关系,但“树叶”只能用OTU的编码表示,rectangular 形状的树形图见图3。
2.4 通过QIIME 和R 语言“ggtree”包优化系统发生树
2.4.1 优化树形 为了使树文件中包含更少的树枝以增加图形的可分辨性,将筛选OTUs 的丰度阈值定为5%。在R 语言运行环境下调用ggtree 包读取筛选后的Newick 格式的树文件rep_set4.tre 绘图,定义树形图的颜色、线条的形状和树形图布局显示物种间进化距离及比例尺、标注内部节点和树枝末端、显示OTUs 编号。根据以上条件绘制的树形图见图4。
2.4.2 向树图中添加物种分类信息 通过QIIME 的assign_ assign_taxonomy.py 脚本将筛选后的OTU 代表序列进行物种注释,即每个OTU 对应一个物种名称,将物种注释文件转换为csv 格式并命名为sample_rep_set4_tax_assignments.csv,需要给文件中的每一列命名,其中第一列“taxa”即为系统发生树文件中的OTU 编号,“taxonomy”列为精确到“种”水平的物种分类信息,最后通过“%<+%”操作符将“taxonomy”列中与OTU 对应的物种分类信息添加到系统发生树中。运行R 代码生成的系统发生树图见图5。

图2 示例样品菌群分类与相对丰度累积条图
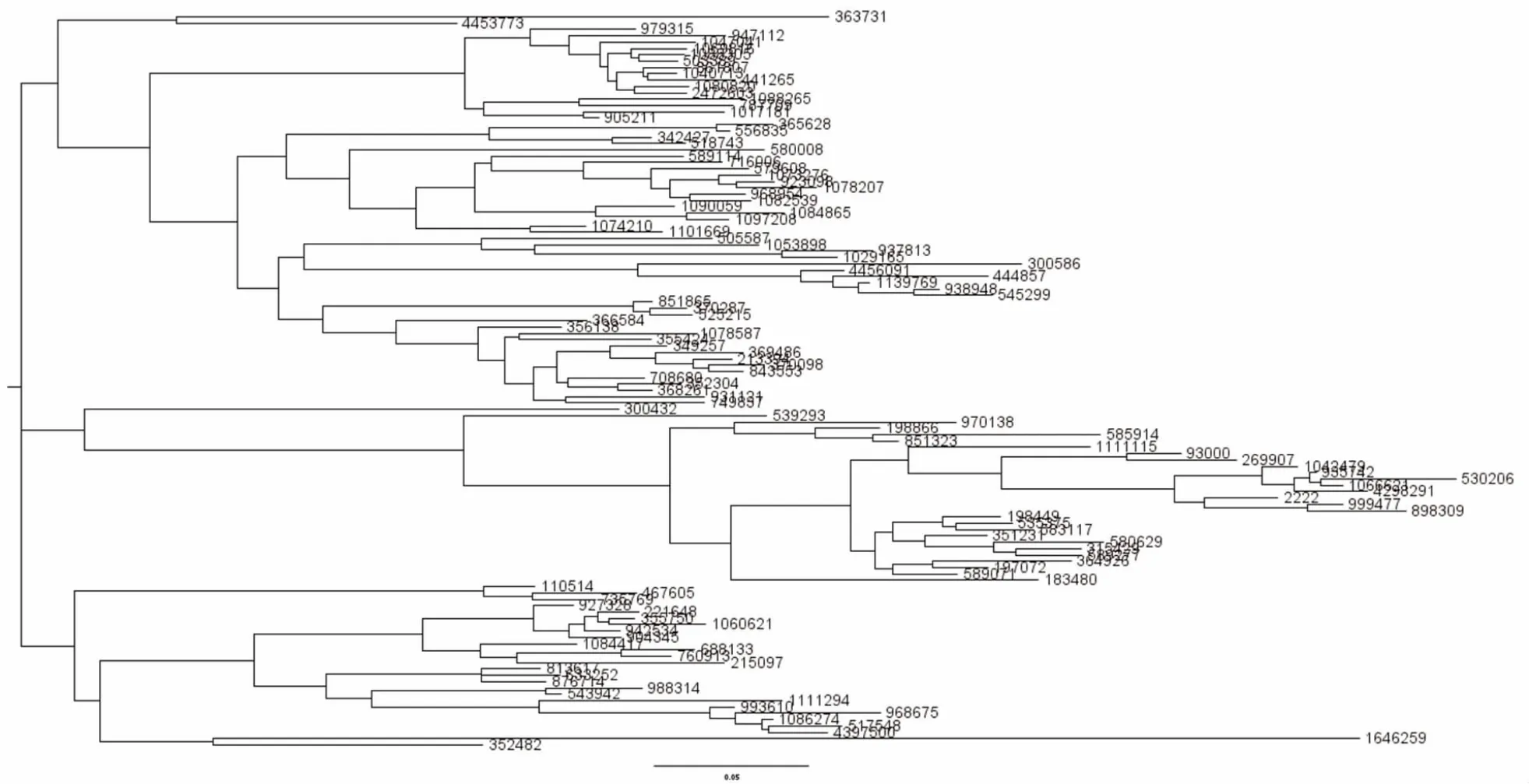
图3 FigTree 软件查看rectangular 布局的原始系统发生树图
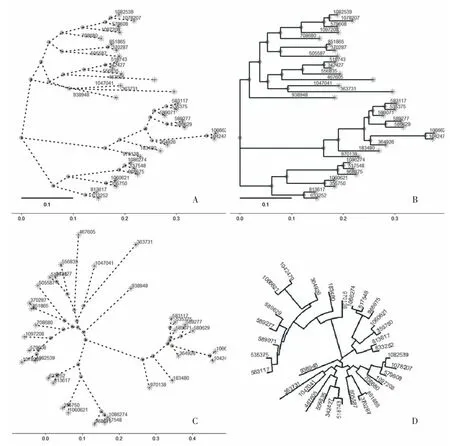
图4 “ggtree”包生成的没有物种注释的系统发生树

图5 ggtree 包绘制的包含物种分类信息注释的系统发生矩形树图
2.4.3 系统发生和物种丰度联合图 通过QIIME 的convert_biom.py 脚本将筛选后的biom 格式的OTUs表文件转换为纯文本格式并命名为table7.from_biom_w_consensuslineage.txt,文件中第一列taxa 即为≥5%丰度的OTU 编号,其它列为每个OTU在不同标本中的丰度(相对含量)值。运行R 语言代码生成的系统发生树与OTU 丰度热图见图6,可以比较方便的查看不同OTU 间进化亲缘关系的远近以及OTU 数量在每个标本中的分布情况。同时,还可以按研究设计的标本分组情况将标本进行合并后作图,对于本示例数据,可以按取样部位将标本合并为肠道(gut)、手掌(palm)和舌部(tongue)来源共3 个组,或者将所有标本合并为1 个组后,重新绘制系统发生和OTU 在不同组间丰度分布的组合图,运行代码生成的系统发生树与OTU 丰度分布热图,结果见图7,其中图7B 所示的联合图系将所有标本合并后绘制系统发生树、点图和条形图的联合图,点的位置和条图的长短均表示相应的OTU 的丰度值。
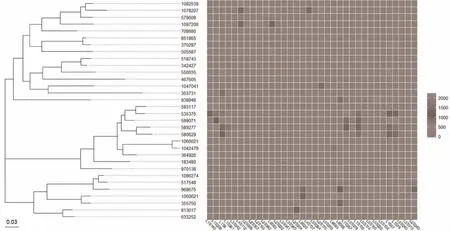
图6 系统发生树和OTU 在不同标本中的丰度分布联合图
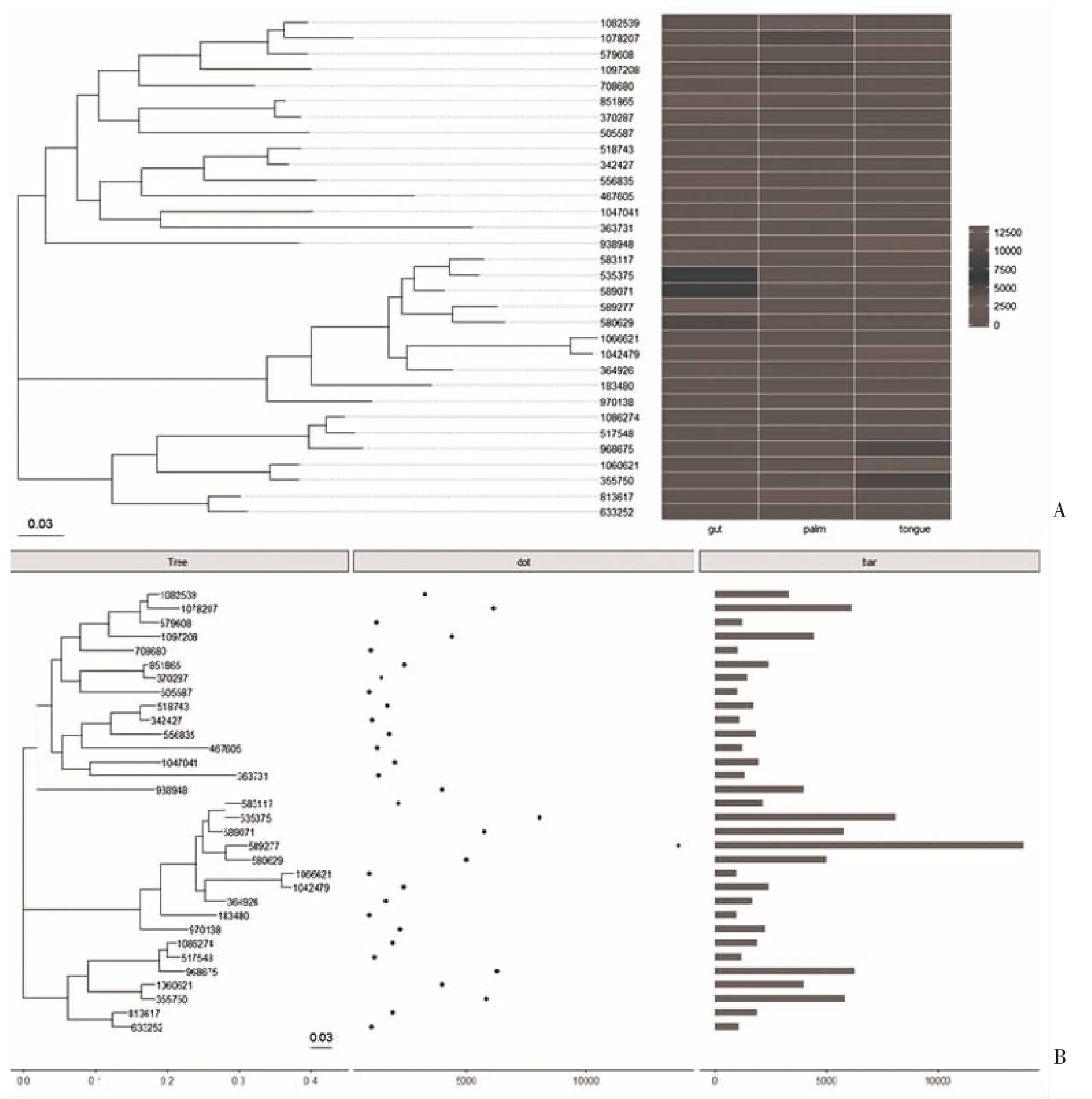
图7 系统发生树和OTU 在不同标本组中的丰度分布联合图
3 讨论
细菌16S rRNA 基因是编码原核生物核糖体小亚基的基因,长度约为1542 bp,在细菌进化过程中的突变率相对较小,并且其分子大小适中,是细菌系统分类学研究中最常用和最有用的分子标志[4]。通过对该基因的V3~V4 单(或双)可变区域进行PCR 扩增和二代测序(NGS),根据测序数据预测标本中微生物群落的种属信息和种属的相对构成比,并进一步研究标本中微生物群落的种属多样性、微生物与环境因子的相互关系,以及微生物功能基因组与宿主和环境因子之间的相互依存关系,这些均是目前微生物学、环境科学和医学研究中的重要课题[5-8]。
用于细菌16S rRNA 基因二代测序数据分析的常用软件主要有 Mothur、QIIME、Uparse、RDP、VAMPS、MEGAN 等,其中Mothur 和QIIME 是最常用的分析工具。QIIME 和R 语言相结合可以解决几乎全部的微生物二代测序数据的整理、统计分析和分析结果的可视化处理。QIIME 的主要用途是运用经过整理后的测序数据,通过聚类分析生成可操作的分类单元(OTUs)数据表明,OTUs 表中的数据为微生物群落的种群分类和数量组成信息。以OTUs表数据作为输入,通过R 语言中的相关程序包可对OTUs 表中的数据进行图形化展示。R 语言作为开源的,面向对象的交互式语言,除了能进行常规的数据处理和统计学分析,近年来有众多学者针对分子生物学实验数据处理的要求开发了大量的数据处理工具,并将这些工具连同部分实验数据以包的形式放置到R 语言环境当中,使用者通过相应代码(命令)调用这些包完成相应的分析任务。在上述分析任务中,通过系统发生树图展示微生物种群之间在种系发生上的相互关系是微生物组数据分析的主要内容。目前多数生物信息学软件生成的微生物组系统树文件只包含最基本的物种间的拓扑关系,结构比较单一。因此本研究设计了基于QIIME 系统和R 语言“ggtree”包的构建系统发生树图操作流程,通过该操作流程,研究者可以向系统发生树图中添加实验设计信息,绘制内容更加丰富的系统发生树图,满足论文发表和研究报告的生成等科研需求。
本工作流程整合了专业分析细菌16S rRNA 基因测序数据的QIIME 系统和用于数据分析和图形化展示的R 语言系统。该方法的特点在于分析过程全部在Windows 系统下进行,其中QIIME 的工作界面虽然是基于linux 系统的,但可以通过VirtualBox在windows 系统中虚拟出一个Linux 系统。并且本流程是基于代码的操作,与其他商业化程序相比具有很高的应用灵活性。更重要的是,工作流程是从原始的测序数据出发的,而不是从由Windows 和菜单用户界面的软件生成的树文件开始,因此可以绘制多种类型的系统发生树形图。使用本流程绘制的图形更准确和美观,最大的优点是能通过各种附加信息对树形图进行多方面的注释,还可以针对系统发生与微生物丰度联合作图。
