白芍非药用部位芍头中单萜苷类有效部位的制备工艺研究
2021-01-19谢凯莉龚慕辛
马 喆,谢凯莉,龚慕辛
白芍非药用部位芍头中单萜苷类有效部位的制备工艺研究
马 喆,谢凯莉,龚慕辛*
首都医科大学中医药学院,北京 100069
优化白芍非药用部位芍头中单萜苷类有效部位的提取及纯化工艺。以芍药苷、芍药内酯苷等单萜苷类成分以及非单萜苷类成分苯甲酸、没食子酸的含量为指标,采用归一化处理得到总评归一值(overall desirability,OD);以提取时间、料液比、乙醇体积分数作为乙醇提取的关键工艺参数,运用星点设计-效应面法建立OD与工艺参数之间的数学模型,推算芍头回流提取的最佳工艺,并进行验证。在单因素试验确定大孔树脂型号的基础上,以上样体积流量、上样液质量浓度、柱径高比、洗脱体积流量作为纯化的关键工艺参数,采用可考察交互作用的正交设计实验优化提取液纯化工艺,并进行验证。芍头回流提取的优选工艺为加12倍量50%乙醇提取2次,每次80 min;3次工艺验证的OD为2.61、2.49、2.57,RSD为2.5%。以D-101型大孔吸附树脂纯化,0.25 g/mL药液上样,上样体积流量为0.5 mL/min,径高比为1∶4,最大上样量为3 BV,2 BV去离子水除杂,4 BV 50%乙醇以1 mL/min体积流量洗脱。3次纯化工艺验证综合评分分别为0.733 3、0.702 4、0.729 7,RSD为2.3%;单萜苷总转移率为74.17%,纯化物中单萜苷质量分数为59.85%。得到的提取和纯化工艺高效且稳定,可除去芍头中大部分非单萜苷成分,获得单萜苷类有效部位。
芍头;星点设计-效应面法;正交设计;提取工艺;纯化工艺;大孔树脂;单萜苷类;没食子酸;氧化芍药苷;芍药内酯苷;芍药苷;没食子酰芍药苷;苯甲酸;苯甲酰芍药苷;归一化
白芍为常用大宗药材,为毛茛科芍药属植物芍药Pall.的干燥根,主要含有芍药苷和芍药内酯苷等单萜及其苷类成分、齐墩果酸等三萜及其苷类、鞣质类、黄酮类、多糖类、挥发油类等[1]。白芍以主根入药,其根茎和侧根凸头俗称“芍头”,在秋季采收时常常作为下脚料被削去,属于非药用部位,干芍头的年产量保守估计有1800~3100 t。已有研究发现芍头中的芍药苷含量远高于主根[2],且与白芍有效成分的种类没有明显差异[3]。据课题组前期调研,目前有将芍头药用或代替白芍提取有效成分的做法。为充分利用芍药非药用部位,促进中药资源合理、合规的使用,针对芍头中单萜苷类成分为主的有效成分进行提取和纯化工艺优化与验证,以达到尽可能保留该类有效成分,去除非单萜苷类成分的目的。
1 仪器与材料
1.1 仪器
1260高效液相色谱仪,美国安捷伦公司;MSA124S-1CE-DU电子天平,赛多利斯科学仪器有限公司;KQ 5200DE型超声波清洗器,昆山市超声仪器有限公司;FW 100型粉碎机,天津市泰斯特仪器有限公司;J1811252旋转蒸发仪,北京西冲科技发展有限公司。
1.2 材料
芍头采集于白芍主产区安徽亳州井泉药业白芍种植基地,经首都医科大学中医药学院罗容副教授鉴定为毛茛科芍药属植物芍药Pall.的干燥根茎。对照品没食子酸(批号BD09A038,质量分数≥98%)、氧化芍药苷(批号A08A0177,质量分数≥98%)、没食子酰芍药苷(批号AF71101-05,质量分数≥98%)、苯甲酰芍药苷(批号AF71101-02,质量分数≥98%)、芍药苷(批号AF71101-01,质量分数≥98%)购自成都普思生物科技股份有限公司;对照品芍药内酯苷(批号S-011-180908,质量分数≥98%)购自成都瑞芬思生物科技有限公司;对照品苯甲酸(批号Z18S7H21155,质量分数≥98%)购自上海源叶生物科技有限公司。
D-101型大孔树脂(批号120613)、AB-8型大孔吸附树脂(批号140611)购自天津市海光化工有限公司;HP-20大孔树脂(批号20190518)购自安徽三星树脂有限公司;HPD-500型大孔树脂(批号M0058-500)、NKA-9型大孔树脂(批号524D011)、S-8型大孔树脂(批号313A011)均购自北京虹湖联合化工产品有限公司;乙腈、甲醇为色谱纯,购自Fisher公司;磷酸为分析纯,购自北京化工厂;95%药用乙醇购自宏发化工有限公司;纯净水购自杭州娃哈哈集团有限公司。
2 方法与结果
2.1 HPLC法同时测定芍头提取物中7种成分含量
2.1.1 混合对照品溶液的制备 精密称取没食子酸、氧化芍药苷、芍药内酯苷、芍药苷、没食子酰芍药苷、苯甲酸、苯甲酰芍药苷7种对照品适量,用甲醇溶解并定容,得到含没食子酸103.0 μg/mL、氧化芍药苷48.5 μg/mL、芍药内酯苷368.0 μg/mL、芍药苷1 122.8 μg/mL、没食子酰芍药苷44.4 μg/mL、苯甲酸41.6 μg/mL、苯甲酰芍药苷54.0 μg/mL的混合对照品母液,依次稀释得到不同质量浓度的混合对照品溶液。
2.1.2 供试品溶液的制备 精密称取芍头最粗粉(过10目筛)25 g,置于500 mL圆底烧瓶中,按照星点设计不同方案进行提取,抽滤并在60 ℃水浴下减压浓缩至无醇味,加相应的提取溶剂转移并定容至生药质量浓度1 g/mL,再稀释得到生药质量浓度0.01 g/mL溶液,0.45 μm微孔滤膜滤过,取续滤液,即得供试品溶液。
2.1.3 色谱条件 色谱柱为Kromasil 100-5 C18柱(250 mm×4.6 mm,5 μm),流动相为乙腈-0.1%磷酸水溶液,梯度洗脱:0~10 min,5%~13%乙腈;10~25 min,13%~15%乙腈;25~40 min,15%~18%乙腈;40~45 min,18%~20%乙腈;45~50 min,20%~50%乙腈;50~55 min,50%乙腈;55~60 min,50%~100%乙腈;60~65 min,100%乙腈;体积流量1.0 mL/min,检测波长:0~10 min,275 nm;10~18 min,258 nm;18~47 min,230 nm;47~50 min,280 nm;50~65 min,230 nm;柱温为25 ℃,进样量为10 μL。理论塔板数按芍药苷计算不低于2000。混合对照品与样品色谱图见图1。
1-没食子酸 2-氧化芍药苷 3-芍药内酯苷 4-芍药苷 5-没食子酰芍药苷 6-苯甲酸 7-苯甲酰芍药苷
1-gallic acid 2-oxypaeoniflorin 3-albiflorin 4-paeoniflorin 5-galloylpaeoniflorin 6-benzoic acid 7-benzoylpaeoniflorin
图1 混合对照品(A) 与芍头样品(B) 的HPLC图
Fig. 1 HPLC of mixed reference substances (A) and rhizome ofsample (B)
2.1.4 标准曲线及线性范围 按“2.1.3”项下色谱条件测定,以各成分色谱峰峰面积作为纵坐标(),对照品中各成分的质量浓度作为横坐标(),绘制标准曲线,计算得回归方程,结果见表1。表明各成分在各自线性范围呈良好线性关系。
2.1.5 精密度试验 将混合对照品母液稀释2倍,精密吸取稀释后的混合对照品溶液10 μL,连续进样6次,按“2.1.3”项下色谱条件测定,并计算各成分峰面积的RSD值,没食子酸、氧化芍药苷、芍药内酯苷、芍药苷、没食子酰芍药苷、苯甲酸、苯甲酰芍药苷峰面积的RSD值分别为0.26%、0.27%、0.22%、0.38%、0.20%、0.40%、0.35%,各成分峰面积的RSD均小于2.0%,表明仪器精密度良好。
2.1.6 重复性试验 精密称取同一芍头样品6份,分别照“2.1.2”项下方法制备供试品溶液,按“2.1.3”项下色谱条件测定,计算各成分质量分数的RSD值,没食子酸、氧化芍药苷、芍药内酯苷、芍药苷、没食子酰芍药苷、苯甲酸、苯甲酰芍药苷峰面积的RSD值分别为1.4%、2.7%、2.6%、2.7%、3.0%、2.3%、2.9%,各成分质量分数的RSD均小于3%,表明该方法重复性良好。
2.1.7 稳定性试验 取同一供试品溶液,分别按“2.1.3”项下色谱条件于0、2、4、6、8、12、24、36、48 h进样,计算各成分峰面积的RSD值,没食子酸、氧化芍药苷、芍药内酯苷、芍药苷、没食子酰芍药苷、苯甲酸、苯甲酰芍药苷峰面积的RSD值分别为0.97%、2.73%、0.30%、0.73%、0.89%、0.48%、0.35%,各成分在48 h内峰面积的RSD均小于3%,表明供试品溶液在48 h内稳定。
2.1.8 加样回收率试验 精密称定1.25 g样品粉末6份,分别精密加入与样品中各成分含量相等的对照品,按“2.1.2”项下方法提取并定容,0.45 μm微孔滤膜滤过,续滤液按“2.1.3”项下色谱条件测定,计算得出7种成分没食子酸、氧化芍药苷、芍药内酯苷、芍药苷、没食子酸芍药苷、苯甲酸、苯甲酰芍药苷的平均加样回收率分别为101.6%、103.0%、99.3%、102.3%、126.7%、102.3%、98.68%,RSD分别为1.3%、1.2%、2.7%、1.8%、1.5%、1.6%、1.8%,均符合要求。
2.2 芍头中各成分含量的测定
精密称取4份芍头细粉(过60目筛)约0.1 g,分别加入10%、30%、50%、70%乙醇50 mL,称定质量,回流提取1 h,冷却后补足质量,过0.45 μm微孔滤膜滤过,取续滤液,按“2.1.3”项下色谱条件测定,计算各成分含量。由于不同体积分数乙醇提取液中各成分含量不同,取最高值计算芍头中各成分含量。
测得实验用芍头中各成分的质量分数分别为没食子酸8.110 mg/g、氧化芍药苷1.044 mg/g、芍药内酯苷10.31 mg/g、芍药苷42.24 mg/g、没食子酰芍药苷3.061 mg/g、苯甲酸2.121 mg/g、苯甲酰芍药苷1.446 mg/g。
2.3 提取工艺的优化
2.3.1 单因素预实验研究 结合白芍提取的相关文献结果[4-12],确定对提取方法及是否粉碎进行考察,并以提取次数、提取时间、料液比、溶剂乙醇体积分数作为单因素实验考察因素,对各因素范围进行考察。结果发现,采用乙醇回流提取时,单萜苷类成分提取率远高于水回流提取及乙醇超声提取,且粉碎后各成分提取率明显优于不粉碎时的提取率。提取2次时提取效果优于提取1次和3次的效果;提取时间为1.5、2.0 h时的提取效果相近且优于提取1.0、2.5 h;料液比为1∶6和1∶10时的提取效果相近且略优于1∶14时,料液比为1∶14时提取效果明显优于1∶20,料液比小于1∶6时溶剂体积难以覆盖药物;70%乙醇提取效果略高于50%乙醇,且两者效果优于40%、60%、80%乙醇,考虑到乙醇体积分数较高会明显增加大生产时的提取成本,因此后续主要研究较低体积分数乙醇的提取工艺。
2.3.2 星点设计试验研究 在前期单因素实验的基础上确定芍头粉粹过10目及乙醇回流提取2次。同时确定了提取时间(1)、料液比(2)、乙醇体积分数(3)3个关键因素,提取时间的极值确定为1 h和2.5 h,料液比的极值确定为1∶6和1∶14,乙醇体积分数的极值确定为30%和60%,因素和水平见表2,提取方法见“2.1.2”项。
2.3.3 星点设计及试验结果 以7种成分的含量()作为指标,采用Hassan法对各正向成分(氧化芍药苷、芍药内酯苷、芍药苷、没食子酰芍药苷、苯甲酰芍药苷)以及负向成分(没食子酸和苯甲酸)的提取率进行归一化处理得到归一值(min)和(max)[13]。
=/
min=(max-Y)/(max-min)
max=(Y-min)/(max-min)
为芍头中各成分含量(mg/g),为供试品溶液中各成分的质量浓度(mg/mL),为稀释倍数,为样品溶液体积(mL),为芍头粉末质量(g)
根据芍头提取物中各成分的含量比例进行主观赋权,各成分权重系数()依次为没食子酸=0.075,氧化芍药苷=0.05,芍药内酯苷=0.20,芍药苷=0.50,没食子酰芍药苷=0.05,苯甲酸=0.075,苯甲酰芍药苷=0.05。将各成分归一值与相应权重系数乘积相加得到总评归一值(OD),星点设计实验与结果见表3。
OD=0.075没食子酸+0.050氧化芍药苷+0.200芍药内酯苷+0.500芍药苷+0.050没食子酰芍药苷+0.075苯甲酸+0.050苯甲酰芍药苷
2.3.4 模型拟合 采用Design-expert软件对数据进行分析得到拟合方程及效应图,拟合方程:OD=2.36-0.0421+0.212+0.403+0.01712+0.01013+0.06623-0.02312-0.1622-0.2532。对模型进行方差分析,结果见表4,效应图见图3。模型值<0.005(差异显著),失拟项的>0.05(差异不显著),表明该模型拟合效果较好。预测极大值为1=1.35 h,2=1∶11.79,C=54.00%时,OD最大,为2.61。考虑到实际生产情况,将优化提取条件定为提取时间(1)=80 min,料液比(2)=1∶12,乙醇体积分数(3)=50%。
2.3.5 验证试验 按照“2.3.4”项中的优化条件提取3份25 g芍头,测定7种指标成分含量,并计算OD值,结果见表5。得到OD值分别为2.61、2.49、2.57,RSD为2.5%。实际值与拟合方程得到的OD预测值2.61相比较,两者基本一致,说明所建立的模型拟合度高,预测性良好。
2.4 纯化工艺的优化
2.4.1 上样液的制备 取25 g芍头粗粉,按照“2.3.4”项中的优化条件提取,提取液浓缩后加水定容至相应质量浓度。
2.4.2 大孔树脂型号的筛选 经查阅文献报道[14-21],确定了待筛选树脂的种类:D-101、HP-20、HPD-500、AB-8、NKA-9、S-8型大孔树脂。
(1)静态吸附的考察:将预处理好的各种型号树脂用滤纸快速吸干表面水分,分别称取2.0 g,置于具塞锥形瓶中,加入5 mL去离子水使其湿润,再精密加入10 mL芍头提取液(生药0.25 g/mL),每隔8 h超声5 min,24 h后抽滤,并以100 mL去离子水洗涤树脂,洗涤液与抽滤液合并后定容至250 mL作为吸附后液,按“2.1.3”项下色谱条件测定。向树脂中加入50%乙醇溶液80 mL,每隔8 h超声振动5 min,24 h后抽滤,并用50%乙醇洗涤,抽滤液和洗涤液合并后定容至250 mL即为解吸液,按“2.1.3”项下色谱条件测定。总评分静态吸附率评分与静态解吸率评分之和,静态吸附率评分和静态解吸率评分分别为各成分吸附率、解吸率与前述相应成分权重系数(非单萜苷类成分权重系数为负)乘积之和,公式如下,评分结果见表6。
静态吸附率()=1-11/00
静态解吸率()=22/(00-11)
静态吸附率评分=−0.075没食子酸+0.050氧化芍药苷+0.200芍药内酯苷+0.500芍药苷+0.050没食子酰芍药苷-0.075苯甲酸+0.050苯甲酰芍药苷
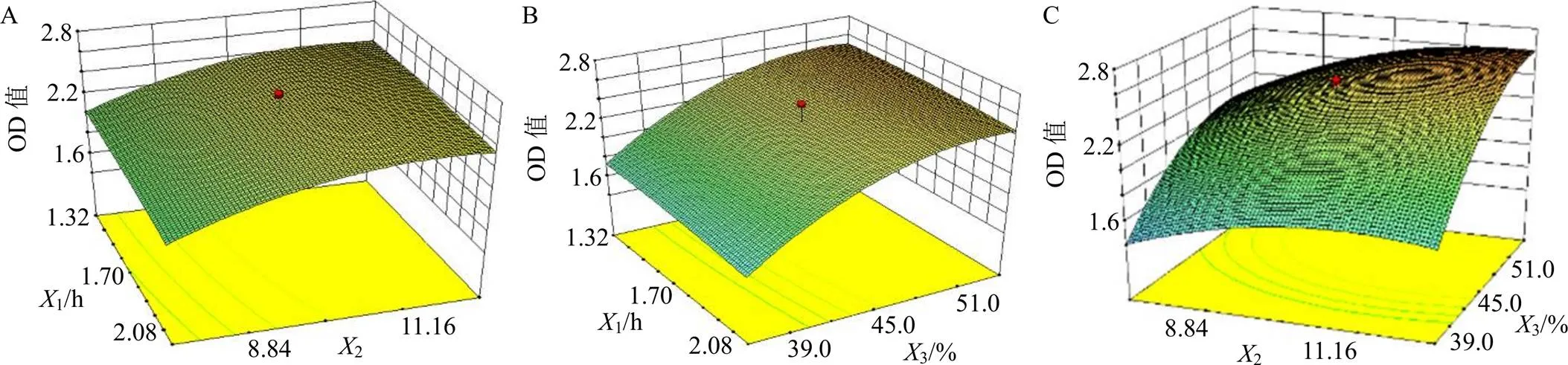
A-提取时间和料液比对提取效果影响 B-提取时间和乙醇体积分数对提取效果影响 C-料液比和乙醇体积分数对提取效果影响

表5 提取工艺验证试验结果
静态解吸率评分=−0.075没食子酸+0.050氧化芍药苷+0.200芍药内酯苷+0.500芍药苷+0.050没食子酰芍药苷-0.075苯甲酸+0.050苯甲酰芍药苷
总评分=静态吸附率评分+静态解吸率评分
0为原溶液中各成分质量浓度,1为吸附后液中各成分质量浓度,2为解吸液中各成分质量浓度,0为原溶液体积,1为吸附后液体积,2为解吸液体积
结果表明,6种树脂中综合评分最高的是非极性大孔树脂D-101,其次是大孔树脂AB-8、HPD-500、HP-20和NKA-9,故淘汰S-8型大孔树脂,采用其他5种树脂进行动态吸附考察。
(2)动态吸附的考察:取同样规格的色谱柱(直径1.5 cm,长30 cm),分别装入上述5种树脂各10 mL,取芍头提取液(生药0.25 g/mL)各20 mL,分别以1 mL/min的体积流量上样,先用50 mL去离子水洗脱,再依次用10%、30%、50%、70%、95%乙醇各30 mL洗脱,洗脱体积流量为1 mL/min,分别收集各部分洗脱液,加去离子水定容至250 mL,其他各部分洗脱液用同浓度乙醇液定容至100 mL,按“2.1.3”项下色谱条件测定。比较5种型号大孔树脂动态洗脱率并进行综合评分,评分方法同“2.3.3”项。结果见表7。
动态吸附率()=(00-22-33)/00
各体积分数乙醇溶液动态洗脱率(W)=CV/00
总洗脱率(总)=10%+30%+50%+70%+95%
0为原溶液中各成分质量浓度,2为上样吸附后流出液中各成分质量浓度,3为水洗脱液中各成分质量浓度,C为各体积分数乙醇洗脱液中各成分质量浓度,0为原溶液体积,2为上样吸附后流出液体积,3为水洗脱液体积,V为各浓度乙醇洗脱液体积
综合考虑动、静态吸附结果,选择D-101、AB-8 2种树脂进行进一步考察。同时发现使用D-101、AB-8 2种树脂,当乙醇体积分数大于50%后,氧化芍药苷、芍药苷、芍药内酯苷、没食子酰芍药苷等大部分成分总洗脱率基本不变,因此后续实验选用50%乙醇进行洗脱。
2.4.3 最大上样量的比较 取同样规格的色谱柱(直径1.5 cm,长30 cm)2根,分别装入D-101、AB-8大孔树脂各10 mL,取芍头提取液(生药0.25 g/mL)分别以1 mL/min的体积流量通过树脂柱,分段收集残留液并定容至25 mL作为吸附后液,前50 mL每10 mL收集1次,以后每5 mL收集1次,按“2.1.3”项下色谱条件测定。发现D-101树脂上样至4 BV时,芍药内酯苷和芍药苷已经开始明显泄露,因此最大上样量为3 BV;AB-8树脂上样至3 BV时,芍药内酯苷和芍药苷已经明显开始泄露,因此最大上样量为2 BV。因此,选择D-101大孔树脂对芍头提取物进行纯化。
2.4.4 纯化工艺单因素预实验研究 结合白芍提取液纯化文献结果,确定以洗脱剂用量、柱径高比、上样液质量浓度、上样体积流量、洗脱体积流量作为单因素实验考察因素,并对各因素范围进行考察。结果发现,为了尽量保存单萜苷类有效成分,除去糖类等杂质,选用20 mL水除杂,40 mL 50%乙醇进行洗脱。柱径高比为1∶4时,纯化效果最好,其吸附率和洗脱率均高于柱径高比为1∶10和1∶1。在一定范围内,上样质量浓度越小时纯化效果越好,但质量浓度过小会导致纯化速度降低,因此后续实验选择0.25、0.5 g/mL。在一定范围内,上样体积流量越小时纯化效果越好,但上样速度过慢过会导致纯化效率降低,因此,确定0.5 mL/min为上样体积流量。在所选择的洗脱体积流量中,当洗脱体积流量为1 mL/min及0.75 mL/min时纯化效果较好。
2.4.5 正交试验 在“2.4.4”项预实验的基础上确定了纯化工艺各影响因素范围的同时,考虑上样液质量浓度(A)与上样体积流量(B)容易有交互作用,当上样液质量浓度增大时,上样速度容易变慢,从而影响上样时间及吸附效果,因此,采用可考察交互作用的正交试验表L8(42)进行优化工艺的筛选,实验安排见表8,重复3次,洗脱率计算公式同“2.4.2”项,综合评分为洗脱率与相应成分权重系数乘积之和,计算方法同“2.3.3”项。
结果见表9,采用SPSS 16.0软件对结果进行方差分析,结果见表10。可以看出,洗脱体积流量(C)和径高比(D)2因素对纯化结果具有显著性影响,因素重要性顺序为C>D>B>A>A×B,最佳上样条件为A1B1C2D2,即当上样质量浓度为生药0.25 g/mL,上样体积流量为0.5 mL/min,洗脱体积流量为1 mL/min,径高比为1∶4时纯化效果最好。
*2=0.464 (adjusted2=0.316)
综合评分=0.075没食子酸+0.050氧化芍药苷+0.200芍药内酯苷+0.500芍药苷+0.050没食子酰芍药苷+0.075苯甲酸+0.050苯甲酰芍药苷
2.4.6 验证实验 按照“2.4.5”项中的最优纯化条件纯化3份芍头提取液,洗脱率计算公式同“3.2.3”项,洗脱率及综合评分见表11,综合评分分别为0.733 3、0.702 4、0.729 7,RSD为2.3%。以每克芍头纯化后所含各成分质量/芍头中各成分的含量计算转移率,芍头中各成分的含量见“2.2”项,3次重复试验的平均转移率分别为没食子酸0.28%、氧化芍药苷36.32%、芍药内酯苷121.95%、芍药苷66.34%、没食子酰芍药35.35%、苯甲酸38.58%、苯甲酰芍药苷71.77%、白芍总单萜苷74.17%。提取物纯化干燥后总单萜苷质量分数为59.85%,苯甲酸质量分数为1.14%,没食子酸质量分数为0.03%。
3 讨论
芍药中的苯甲酸会增加肝脏的解毒负担,含量不宜过高[22];没食子酸为非单萜苷类成分,因此苯甲酸和没食子酸为评价指标中的负向成分。氧化芍药苷、芍药内酯苷、芍药苷、苯甲酰芍药苷为白芍总苷类有效成分[23],没食子酰芍药苷具有抗炎、抗溃疡及免疫调节等作用[24],这5种成分是正向成分。
在预实验中,乙醇体积分数为60%时提取效果较为50%及70%均差,分析原因是指标成分较多,各成分极性不同,因此乙醇体积分数与提取效果的关系呈M型。在提取过程中,当乙醇体积分数低于40%时,会出现大量絮状物质,导致后续过滤困难,提示大生产使用低体积分数乙醇不适宜,同时不宜使用60%乙醇。
考察纯化工艺时,提取液浓缩至无醇味并用水定容至1 g/mL后,久置会产生少许沉淀,摇匀后加水稀释成其他质量浓度后沉淀会减少,上样过程中树脂上层会有少量沉淀,但用乙醇洗脱后沉淀溶解,不影响后续实验的洗脱速度和洗脱时间。
采用优化工艺得到的提取液干浸膏得率为36.8%,单萜苷类成分质量分数为12.86%,因此需要进一步纯化。已有研究中常用来纯化白芍提取物的大孔树脂大多为非极性树脂,更利于除去提取物中的糖类成分。本实验中,采用2 BV水可将提取液中大部分多糖等杂质洗脱,且仅洗脱下少量芍药苷。相比使用50%乙醇,70%乙醇除苯甲酸和苯甲酰芍药苷外对其他5种成分的洗脱率基本不变,而苯甲酸为负向成分,苯甲酰芍药苷的含量远低于芍药内酯苷和芍药苷,因此,为了尽量提高芍药内酯苷和芍药苷的洗脱率并适当降低成本,选择了50%乙醇进行洗脱。文献中白芍提取物的纯化工艺中,通常以芍药苷转移率、白芍总苷转移率、浸膏得率等因素作为评价指标,所得的最优纯化条件,柱径高比为1∶7~1∶10,上样液质量浓度大部分为0.5 g/mL,也有0.1、0.2、1.0 g/mL;上样体积流量为2.4~5 BV/h,洗脱体积流量为2~7 BV/h,也有文献以滴/s作为体积流量单位,换算后上样体积流量和洗脱体积流量分别为9、18 BV/h,和其他文献差异过大。实验过程中,体积流量的调节较难控制,且随着溶液流出,体积流量逐渐减小,这可能是造成最优体积流量差异的原因之一。除此之外,部分文献中将提取液进行离心处理后再进行纯化,也可能是造成实验结果差异的原因。
芍药苷和芍药内酯苷为白芍提取物中含量最高的2种有效成分,两者为同分异构体。有关芍药苷稳定性的研究发现,芍药苷在大于80 ℃、偏碱性环境、水液中、光照情况下可发生降解[25-27],同时芍药苷的半缩酮结构可与溶剂中的乙醇发生缩酮反应导致降解[28]。本研究提取用溶剂为50%乙醇,沸点高于80 ℃,尽管减压回收中温度控制在60 ℃以下,水液不偏碱性,也可能存在芍药苷分解。另有研究发现经过加热炮制后的白芍中的芍药内酯苷含量高于生品[29],与本实验中芍药苷转移率偏低,芍药内酯苷转移率较高甚至超过100%的现象类似。芍药苷和芍药内酯苷在一定条件下可相互转化[30],课题组前期测定白芍花中芍药苷和芍药内酯苷的含量时,发现在3个品种的白芍花中均存在芍药苷含量鲜花中高,干燥花中降低而芍药内酯苷升高的现象[31],认为温度升高可能会促进芍药苷转化为芍药内酯苷,因此应将2种成分的转移率合并判断实验结果。如将目前得到的优化工艺中纯化后超出原药材含量的芍药内酯苷视为芍药苷转化而来,则可计算出芍药苷转移率应大于71.70%。
总之,本研究优化的从芍头中提取总单萜苷有效部位的制备工艺较稳定、可行,为白芍非药用部位的开发提供了参考,降低了白芍单萜苷有效部位提取原料的成本,提高了药农收入,有利于绿色中药种植的可持续发展。
利益冲突 所有作者均声明不存在利益冲突
[1] 吴芳, 杜伟锋, 徐姗姗, 等. 白芍化学成分及质量评价方法研究进展 [J]. 浙江中医药大学学报, 2012, 36(5): 613-615.
[2] 郑金凤, 王景红, 宗志勇, 等. 基于HPLC技术分析白芍根茎与主根部分的主成分含量差异 [J]. 环球中医药, 2016, 9(8): 928-930.
[3] 宗志勇, 郑金凤, 路广义, 等. 白芍药材主根与根茎的HPLC指纹图谱比较研究 [J]. 中国药房, 2017, 28(27): 3865-3867.
[4] 甘恕潮, 李成网. 正交试验法提取白芍中芍药甙的工艺研究 [J]. 淮海医药, 2001, 19(4): 344-345.
[5] 张慧, 付京, 陈杨, 等. 正交试验法优化白金胶囊中白芍的提取工艺 [J]. 中国中医药信息杂志, 2014, 21(7): 64-66.
[6] 侯继秋, 尹寿玉, 柳明洙. 正交实验法优选白芍中芍药苷的醇提工艺 [J]. 时珍国医国药, 2007, 18(3): 657-658.
[7] 何凤兰. 星点设计-效应面法优化白芍提取工艺 [J]. 海峡药学, 2014, 26(8): 23-25.
[8] 魏词, 万蒞, 王红, 等. 星点设计-效应面法优化白芍提取工艺 [J]. 医药导报, 2016, 35(2): 174-176.
[9] 尹雪, 孙萍, 温学森, 等. 混合均匀设计法优化白芍提取工艺 [J]. 中国药房, 2016, 27(1): 89-91.
[10] 陈象青, 屈建, 卢今, 等. 田口实验设计法优选白芍中芍药苷的提取工艺 [J]. 中国新药杂志, 2013, 22(15): 1836-1839.
[11] 杨士友, 孙备, 黄世福, 等. 白芍中白芍苷提取和纯化工艺的研究 [J]. 安徽医药, 2004, 8(1): 21-23.
[12] 贲永光, 李坤平, 李康, 等. 正交设计研究白芍总苷的超声提取工艺 [J]. 食品与生物技术学报, 2009, 28(4): 501-504.
[13] 吴伟, 崔光华, 陆彬. 实验设计中多指标的优化: 星点设计和总评“归一值”的应用 [J]. 中国药学杂志, 2000, 35(8): 530-533.
[14] 陈佳亮, 艾萍, 张金玲, 等. 白芍中芍药苷大孔树脂纯化工艺研究 [J]. 时珍国医国药, 2012, 23(5): 1159-1160.
[15] 陈佳亮.大孔吸附树脂在中药分离纯化应用中影响因素研究 [D]. 北京: 北京工业大学, 2012.
[16] 樊燕, 饶光玲. 大孔吸附树脂分离纯化白芍芍药苷的工艺研究 [J]. 贵阳医学院学报, 2012, 37(1): 30-33, 36.
[17] 郑军献, 浦锦宝, 胡轶娟, 等. 大孔吸附树脂分离纯化白芍总皂苷的研究 [J]. 医学研究杂志, 2010, 39(11): 38-40.
[18] 罗扬. AB-8大孔树脂对白芍提取液的最佳动态纯化条件 [J]. 遵义医学院学报, 2009, 32(2): 183-185.
[19] 蔡一杰, 金传山, 吴德玲, 等. 大孔树脂纯化白芍总苷的研究 [J]. 安徽医药, 2009, 13(3): 245-246.
[20] 薛建国, 狄留庆, 赵晓莉. 白芍中芍药总苷类化合物的提取与分离工艺研究 [J]. 江苏中医药, 2006, 38(12): 55-58.
[21] 张亚军, 徐莲英. HPD100大孔吸附树脂在白芍纯化工艺中的应用研究 [J]. 中药材, 2006, 29(9): 968-970.
[22] 陈科敏, 吴巧凤. 杭白芍和亳白芍中苯甲酸的含量测定 [J]. 中草药, 2004, 35(9): 1064.
[23] 郑琳颖, 潘竞锵, 吕俊华, 等. 白芍总苷药理作用研究 [J]. 广州医药, 2011, 42(3): 66-69.
[24] 侯玮琛.没食子酰芍药苷通过PI3K/Akt/Nrf2通路减轻脑缺血再灌注后炎症反应与氧化应激损伤 [D]. 长春: 吉林大学, 2017.
[25] 霍晓光, 胡欣彤, 陈丽霞, 等. 芍药苷稳定性研究 [J]. 中国科技论文, 2017, 12(18): 2092-2097.
[26] 罗娟, 呼延玲, 刘晓玲. 光照对芍药苷稳定性的影响 [J]. 中国药师, 2011, 14(1): 87-88.
[27] 闫兴丽.乾坤清颗粒制剂工艺、质量标准及芍药苷稳定性的研究 [D]. 北京: 北京中医药大学, 2003.
[28] 唐安玲, 郑琰, 宋英, 等. 芍药苷在疏经防痛胶囊水提液和浓缩液中热稳定性考察 [J]. 中国实验方剂学杂志, 2015, 21(7): 24-26.
[29] 沈梦兰, 严斌俊, 秦路平. 不同产地、炮制方法、采收时间的芍药中有效成分含量差异研究进展 [J]. 浙江中医药大学学报, 2019, 43(6): 622-630.
[30] 刘鑫鑫, 马骁驰, 霍长虹, 等. 芍药苷和芍药内酯苷的微生物转化 [J]. 中国中药杂志, 2010, 35(7): 872-875.
[31] 谢凯莉, 马喆, 龚慕辛, 等. 不同栽培品种及干燥工艺白芍花中9种有效成分含量测定及主成分分析 [J]. 中国中药杂志, 2020, 45(19): 4643-4651.
Study on preparation of monoterphene glycosides from the non-medicinal part-rhizome of
MA Zhe, XIE Kai-li, GONG Mu-xin
School of Traditional Chinese Medicine, Capital Medical University, Beijing 100069, China
To optimize the extraction and purification process of active parts of monoterpene glycosides from the non-medicinal part-rhizome of.The contents of multiple components were used as indexes, including monoterpenoid glycosides, such as paeoniflorin and albiflorin, and non-monoterphene glycosides components, such as benzoic acid and gallic acid. The normalization method was used to obtain the overall desirability (OD). The extraction time, solid-to-liquid ratio and ethanol volume fraction, as the key process parameters, were optimized through central composite design-response surface methodology, and the results were verified. Based on the single factor tests used to determine the type of macroporous resin, the loading volume flow rate, loading solution mass concentration, column diameter to height ratio, and elution volume flow rate were the key process parameters for purification. Orthogonal design experiments that can investigate the interactions were used to optimize the extraction liquid purification process and the results were verified.The optimal reflux extraction process of rhizome ofwas to extract twice with 12 times of 50% ethanol, 80 minutes eachtime. The OD values of three validation experiment were 2.61, 2.49, 2.57, and the RSD was 2.5%. D-101 macroporous adsorption resin was selected for the purification, 0.25 g/mL drug solution was loaded, the sample volume flow rate was 0.5 mL/min, the diameter-to-height ratio was 1: 4, the maximum sample load was 3 BV, 2 BV deionized water was used for impurity removal, and 4 BV 50% ethanol was eluted at the volume flow rate of 1 mL/min. The comprehensive scores of the three purification process verifications were 0.733 3, 0.702 4, 0.729 7, the RSD was 2.3%, and the total transfer rate of monoterpene glycosides was 74.17%, and the mass fraction of monoterpene glycosides in the purified product was 59.85%.The optimum extraction process is efficient and steady, which can remove most of non-monoterpene glycosides in the rhizome of, and obtain the active fraction of the monoterpene glycosides in the rhizome of.
rhizome ofPall.; central composite design-response surface methodology; extraction process; purification process; macroporous resin; monoterpene glycosides;gallic acid;oxypaeoniflorin;albiflorin;paeoniflorin; galloylpaeoniflorin; benzoic acid;benzoylpaeoniflorin;overall desirability
R284.2
A
0253 - 2670(2021)02 - 0386 - 10
10.7501/j.issn.0253-2670.2021.02.011
2020-08-23
国家重点研发计划项目(2017YFC1701903-3)
马 喆(1997—),女,硕士研究生,研究方向为中药学。E-mail: 1144938549@qq.com
龚慕辛(1968—),女,博士,教授,博士生导师,主要从事中药制剂工艺与质量标准研究。Tel: (010)83911624 E-mail: gongmuxin@126.com
[责任编辑 郑礼胜]
