硫掺杂石墨烯电催化降解有机染料甲基橙
2021-01-18陈思胡腾飞于永波王冰鑫洪俊明张倩
陈思,胡腾飞,于永波,王冰鑫,洪俊明,张倩
(1 华侨大学化工学院,福建厦门361021;2 福建省工业废水生化处理工程技术研究中心,福建厦门361021)
伴随着我国印染行业工艺技术的不断发展,工业染料废水的排放量也随之日益增长。工业染料废水具有成分复杂、有机物含量高、色度高、酸碱性强并且生物毒性高等特点[1],随意排放将会对人类及自然界造成极大的危害。常用于处理工业染料废水的方法有絮凝法、吸附法、生物氧化法、光催化法、化学氧化法等[2]。其中,电催化氧化法利用在阳极产生的活性自由基氧化降解有机分子,具有操作简单、条件温和、无二次污染、能源利用率高、可重复利用、易实现自动化等特点[3],在实际应用中具有良好的发展前景。
电催化氧化法的降解效果主要受到阳极材料的影响。与常用的高成本贵金属材料相比,石墨烯材料凭借着高比表面积、优良的导电性能以及强机械性能,成为制备阳极材料的一个新的选项[4]。纯石墨烯薄片易团聚,会丧失其在薄层状态下的优良性能,因此常采用化学掺杂法对石墨烯进行改性[5]。硫的原子半径较碳大,硫的引入会带来石墨烯非平面结构杂化的改变,并产生有利于电子吸附的结构空隙,从而提高电化学性能[6],与碳相似的电负性常被用于掺杂在石墨烯中。硫掺杂后带来的电荷分布变化以及形成的C—S—C键和C—SOx—C键等结构能够有效增强材料的氧化还原能力,有利于材料催化性能的提高[7]。
与氮原子和硼原子相比,将硫原子结合到石墨烯中需要更高的能量,因此硫掺杂石墨烯的合成更加困难[8]。Jeon 等[9]用原始石墨和硫(S8)采用球磨法合成边缘硫化石墨烯,硫含量达到4.9%左右。球磨法可以批量制备硫掺杂石墨烯,但无法对掺杂含量等方面进行精准有效的调控[10]。Gao 等[11]以硫粉和正己烷为原料,在氢氩混合气体中通过化学气相沉积法(CVD)制备出硫含量仅为0.6%的硫掺杂石墨烯薄膜。由于CVD 沉积法常以金属作为催化剂,因此在研究硫掺杂石墨烯性能时还需考虑金属元素引入带来的影响[12]。而高温热退火法是一种广泛应用于石墨烯掺杂硫的方法,具有简单可行易调控等优势[8]。Yang等[13]以氧化石墨烯(GO)和二苄基二硫为原料,在氩气中采用高温热退火法成功合成硫含量约为1.5%且含有C—S—C 和C—SOx—C 结构的硫掺杂石墨烯,其电催化活性优于商用铂碳催化剂。目前,关于硫掺杂石墨烯大部分集中在超级电容器、ORR 以及储能电池等方面,应用于电催化降解的还鲜有报道。
本文采用改进高温热退火法,将常用的惰性气体替换为高还原性气体一氧化碳(CO),合成掺杂量较高的硫掺杂石墨烯作为电催化阳极材料,降解有机染料甲基橙废水,探究了硫掺杂石墨烯的微观结构特征和表面官能基团。对比硫掺杂石墨烯与还原石墨烯对污染物的处理效率,考察了电解质、初始电流、甲基橙浓度、初始pH 以及实验温度等因素对降解过程的影响,探究硫掺杂石墨烯电催化降解有机污染物的运行条件,为硫掺杂石墨烯电催化技术在难降解有机废水处理的应用提供参考。
1 材料与方法
1.1 实验试剂
石墨粉(约30μm)、硝酸钠(NaNO3)、高锰酸钾(KMnO4)、过氧化氢(H2O2,30%)、盐酸(HCl)、氯化钠(NaCl)、甲基橙(methyl orange),国药集团化学试剂有限公司;硫酸(H2SO4,98%)、甲醇(CH3OH,99%)、乙醇(C2H5OH,99.7%)、二苯二硫醚(PDS),西陇科技有限公司。所用试剂均为分析纯,溶液均用去离子水配制。
1.2 氧化石墨烯(GO)制备
氧化石墨烯的制备采用改进的Hummers[14]方法,主要制备过程可以分为低温、中温和高温共三个阶段。
低温阶段:将石墨粉(1g)、硝酸钠(1g)和硫酸(98%,50mL)在圆底烧瓶中混合,于冰浴条件下(0℃)缓慢加入高锰酸钾(6g),保持温度在5℃以下,磁力搅拌器搅拌30min 至溶液呈青绿色。
中温阶段:向反应器中缓慢滴加去离子水(10mL),提升反应温度至35~45℃,持续搅拌30min。
高温阶段:缓慢滴加去离子水(100mL)使混合液温度保持在95℃,持续搅拌直至溶液呈深棕色后加入适量30%过氧化氢(15mL)终止反应。将混合物冷却静置后,加入稀盐酸(1mol/L,100mL)清洗离心1 次,所得下层沉淀再用乙醇(100mL)清洗离心2 次,纯化获得氧化石墨。将氧化石墨用乙醇稀释至100mL,得到棕黄色氧化石墨悬浮液。
将氧化石墨悬浮液置于超声波清洗器中,水浴超声(超声功率100W,超声温度25℃)1h后得到氧化石墨烯分散液,保存备用。
1.3 硫掺杂石墨烯(SGN)及还原石墨烯(RGN)的制备
将GO 分散液(5mL,15g/L)和二苯二硫醚(PDS)(0.1g) 分散于20mL 甲醇溶液中,超声5min 中后形成均匀的混合液体。将所得混合液置于40℃烘箱中干燥过夜,得到固体混合物。将混合物放置于具有CO 氛围的石英管中,并在600℃下加热2h,冷却后取出。为进行对比实验,同样将GO分散液(5mL)分散在20mL甲醇中,采取与上述相同方法制备还原石墨烯。
1.4 电催化性能测试
构建三电极反应系统进行自制材料电催化性能测试。称取4mg SGN材料,用导电胶将材料均匀涂布在大小为1.5cm×1.5cm 的洁净碳布上,干燥后作为阳极。以10cm×1.5cm 的铜片和Ag/AgCl 电极为阴极和参比电极。调节极板间距为1cm,利用PS−12阳极极化仪(欧亚中兴技术)控制外加电流为10~40mA,甲基橙溶液200mL,考察电解质种类(Na2SO4、NaNO3、NaCl)、外加电流(10mA、20mA、30mA、40mA)、甲基橙浓度(10mg/L、20mg/L、30mg/L)、初始pH(3、5、7、9、11)以及反应温度(20℃、35℃、45℃)对于催化效率的影响。染料浓度测定采用TU−1900 双光束紫外可见分光光度计,在458nm 波长下测定。并通过式(1)计算其降解率。

式中,C 为t 时刻甲基橙浓度;C0为甲基橙初始浓度;A 为t 时刻甲基橙的吸光度;A0为甲基橙初始吸光度。
1.5 材料表征
催化剂的表面形貌通过扫描电镜(SEM)图像(Tescan Mira3 Zeiss Sigma500) 和透射电镜(TEM)图像(JEOL−2100F)进行观察;利用傅里叶变换红外光谱(FTIR,Thermo Scientific)分析材料表面的官能基团;采用拉曼光谱仪(Raman,Renishaw in Via) 分析碳材料表面缺陷;利用元素分析仪(Elementar, Vario MICRO Cube)分析材料元素含量;采用ESCALAB250Xi PHI 单色Al Kα辐射(1486.6eV)的X 射线光电子能谱(XPS,ESCALAB250XiX) 分析材料内部键合。
2 结果与讨论
2.1 材料表征
2.1.1 SEM与TEM分析

图1 材料的扫描电子显微镜与透射电子显微镜图
利用扫描电子显微镜图和透射电子显微镜图对材料的表观和微观形貌特征进行测定,结果如图1所示。从SEM 图中可以看出制备的SGN 材料具有良好的片层结构,表明石墨烯剥离较为成功。同样,在TEM 图中也有映照。将SGN 与RGN 的TEM图进行对比,可以看出两种材料均呈透明褶皱薄纱状,表面较为平整,但SGN 材料存在一定的堆叠情况,并且片层厚度明显增加。这主要是由于硫原子半径较大,掺杂后引起石墨烯键长和键角的变化,导致片状褶皱和堆叠结构的形成,并使片层增厚[15]。
2.1.2 红外图谱分析
对比SGN 和RGN 的红外光图谱(图2),进行材料表面官能团和化学键分析。SGN 和RGN 图谱中 均 在3435cm−1和1630cm−1处 出 现 明 显 的H2O的—OH 伸缩振动峰和C==C 伸缩振动[16]响应。SGN中 在1230cm−1、1120cm−1、1020cm−1处 有 明 显 的S==O 的伸缩振动和S−phenyl 的伸缩振动[17],而在618cm−1处存在明显的C—S 伸缩振动峰[18]。观察图2可知,RGN与SGN的含氧官能团较少,可能是因为在高温CO 氛围下,含氧官能团大部分被还原。与RGN 相比,SGN 中1630cm−1处C==C 伸缩振动峰的吸收强度明显减小,可能是由于硫掺杂导致部分C=C 的破坏,使其含量相应减少[16]。而SGN 中S==O 和C—S 伸缩振动峰的吸收强度明显强于RGN,证实SGN中硫的成功掺杂。

图2 硫掺杂石墨烯和还原石墨烯的红外光谱
2.1.3 拉曼光谱分析
用拉曼光谱对材料中碳的晶体缺陷进行分析,所得结果如图3 所示。在石墨烯的拉曼光谱图中,1350cm−1附近为D带,表示缺陷的存在,揭示碳原子无序排列的程度[19]。1580cm−1附近为G 带,对应于碳原子sp2杂化轨道的伸缩振动[20]。因此,D峰与G 峰的强度比值(ID/IG)可以反映出碳材料的缺陷程度。根据计算可得还原石墨烯的ID/IG值(0.9057)大于硫掺杂石墨烯的ID/IG值(0.8249),表明还原石墨烯的缺陷程度更大,可能是由于硫掺杂石墨烯材料引入了六环碳结构,导致sp2杂化碳结构的增加[20]。
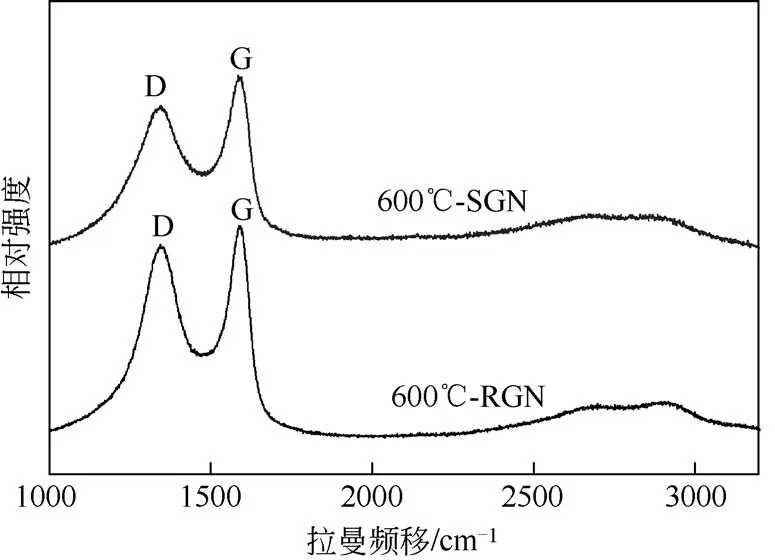
图3 硫掺杂石墨烯和还原石墨烯的拉曼光谱图
2.1.4 元素分析
利用元素分析仪对GO、RGN 和SGN三种材料进行碳、氧、硫元素的含量分析,结果见表1。由表中信息可知,与GO相比,RGN和SGN的氧元素含量明显降低,进一步佐证GO的含氧官能团被还原,与红外光谱图结果相对应;SGN中硫元素含量高达11.2%,与文献[7−8]中报道的含量相比较高,推测可能是由于高温CO 的环境下促进了硫原子的掺杂。

表1 不同材料中碳、氧、硫元素的测定结果(原子分数)
2.1.5 XPS图谱分析
利用X射线光电子能谱对SGN的元素组成和化学价态进行分析。如图4(a)X 射线光电子能谱总谱图所示,SGN存在S 2p(168.4eV)、S 2s(230eV)、C 1s(284.9eV)和O 1s(531eV)四个特征峰。通过计算可知S、C、O 元素的原子含量分别为10.29%、56.57%、32.74%,该材料表面因羟基和羧基等官能基团的存在还含有少量H元素。本研究中自制材料中硫原子含量高达10.29%,与元素分析仪所测结果相近。C 1s的谱图如图4(b)所示,图中284.5eV 处的特征峰对应于sp2杂化碳,285.3eV处的特征峰对应C—O/C—S,而285.9eV 处的特征峰 则 对 应C—OH[21−22]。图4(c)为O1s 光 谱 图,529.6eV和531.6eV分别与C—O官能团和S==O相对应[23−24]。由S2p光谱图[图4(d)]可知,硫掺杂主要以C—S(164.2eV)和C—SOx—C(168.9eV)两种形式存在于材料当中, 并含有少量C==S(165.5eV)结构[25−26]。根据XPS 谱图分析可知,硫成功掺杂至石墨烯中,并且与红外谱图和拉曼光谱图相对照,证实了S==O、C—S和sp2杂化碳等结构的存在。

图4 硫掺杂石墨烯的X射线光电子能谱图
2.2 催化性能测试
2.2.1 材料吸附性能影响
由于RGN 和SGN 对染料具有一定的吸附效果[8,21],为了排除实验过程中材料吸附性能带来的影响,在初始pH 为7、NaCl 为电解质、甲基橙浓度为10mg/L 且实验温度为20℃的条件下,对RGN和SGN 材料的吸附性能进行实验,结果如图5 所示。根据图5可知,RGN和SGN材料对甲基橙染料几乎没有任何吸附效果。这可能是因为与200mL的甲基橙溶液相比,RGN 和SGN 材料用量较少,仅为4mg,所以在后续实验过程中可以忽略RGN和SGN材料的吸附性能影响。

图5 硫掺杂石墨烯和还原石墨烯吸附性能
2.2.2 电解质的影响
在初始pH 为7、外加电流为20mA、甲基橙浓度为10mg/L 且实验温度为20℃的条件下,对Na2SO4、NaNO3、NaCl 三种不同的电解质进行研究。由图6可知,选用NaCl为电解质时,降解速率最快,在60min 时降解率已达到94.14%;而选用Na2SO4和NaNO3作为电解质时,在120min时降解率仅为28.75%和19.70%。NaCl 作为电解质时降解效率最佳,主要原因是Cl−可通过电催化产生活性氯[式(2)],并进一步生成具有强氧化性的ClO−[式(3)、式(4)],从而促进降解甲基橙[14,27]。

2.2.3 外加电流的影响

图6 不同电解质对硫掺杂石墨烯电催化降解甲基橙的影响

图7 不同外加电流对硫掺杂石墨烯电催化降解甲基橙的影响
在初始pH 为7、甲基橙浓度为10mg/L、电解质为NaCl 且实验温度为20℃的条件下,研究不同外加电流对硫掺杂石墨烯电催化降解甲基橙的影响。如图7所示,当外加电流从10mA增加到20mA和30mA时,硫掺杂石墨烯的降解效率明显提升。主要是由于电流的增大会加速电子的产生,并使得电子传递速度的增加,从而提高电催化降解的效率[28]。但当电流从30mA 增加到40mA 时,硫掺杂石墨烯的降解效率略有下降。这可能是由于在过大的外加电流下电极发生副反应,由原来的生成羟基自由基[式(5)]变为生成氧气[式(6)],从而影响电催化降解效率[29]。因此,选用30mA 电流密度的外加电流。

2.2.4 甲基橙浓度的影响
在初始pH 为7、外加电流为30mA、电解质为NaCl且实验温度为20℃的条件下,研究不同浓度的甲基橙对电催化降解效果的影响。从图8中可以看出,浓度越高的甲基橙所需降解时间越长,10mg/L、20mg/L和30mg/L的甲基橙降解率达到95%左右分别需要45min、60min和70min。由于单位时间内SGN可产生的羟基自由基数量有限,所以降解更高浓度的甲基橙则需耗费更多时间[27]。因此,在后续影响因素研究中采用10mg/L的甲基橙作为降解浓度。
2.2.5 初始pH的影响

图8 硫掺杂石墨烯降解不同浓度甲基橙
在甲基橙浓度10mg/L、外加电流为30mA、电解质为NaCl 且实验温度为20℃的条件下,研究不同初始pH 对硫掺杂石墨烯电催化降解甲基橙的影响。如图9 所示,随着pH 的增加,硫掺杂石墨烯的电催化降解效率不断降低。其中降解效果最佳的条件为初始pH 为3 时,在35min 时降解率即达到98.49%。而当初始pH 增大至11 时,60min 时降解率仅为84.14%。首先,在酸性条件下,阳极的析氧电位较高,有利于羟基自由基的生成,因此降解效率更高[30]。其次,甲基橙在pH 为3 时为醌式结构,而在碱性条件下为偶氮结构[31]。醌式结构比偶氮结构更容易氧化降解,因此初始pH 为3 时的降解效果最好[32]。在碱性条件下,析氧反应阻碍了羟基自由基的生成,且羟基自由基容易与OH−反应生成氧化性较弱的·O−,影响甲基橙的降解效率[30]。

图9 不同初始pH对硫掺杂石墨烯电催化降解甲基橙的影响
2.2.6 反应温度的影响
在初始pH 为7、甲基橙浓度为10mg/L、电解质为NaCl且外加电流为30mA时,对硫掺杂石墨烯分别在实验温度为20℃、35℃和45℃电催化降解甲基橙的降解效率进行了研究。如图10 所示,当实验温度从20℃提升到35℃和45℃时,甲基橙的降解率达到95%,用时从40min 缩减至30min,反应速率有一定的提升。

图10 温度对硫掺杂石墨烯电催化降解甲基橙的影响
2.2.7 电极材料的稳定性
为测定硫掺杂石墨烯作为电极材料的稳定性和可重复使用性,在初始pH 为7、甲基橙浓度为10mg/L、电解质为NaCl、外加电流为30mA且实验温度为20℃的条件下,进行电极材料稳定性实验。结果如图11 所示,将涂覆SGN 的电极在相同条件下,重复实验5次,降解率仍未发生明显改变,均达到95%以上。实验结果表明,电极材料的稳定性良好,可重复使用。

图11 硫掺杂石墨烯重复使用实验
2.2.8 硫掺杂石墨烯的电催化性能
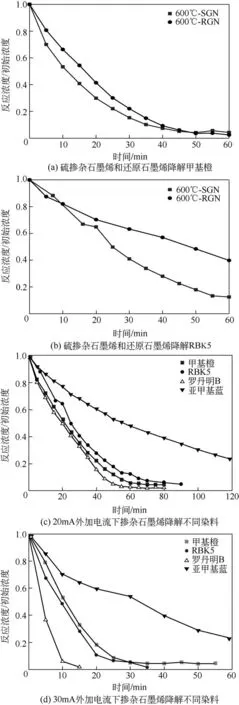
图12 硫掺杂石墨烯的电催化性能
在初始pH 为7、外加电流为30mA、电解质为NaCl、实验温度为20℃条件下,利用RGN 和SGN对甲基橙溶液(10mg/L)进行降解,对比研究材料的电催化性能,结果如图12(a)所示。结果表明,RGN和SGN对甲基橙均有良好的降解效果,50min内降解率可高达95%,其中SGN 前期的降解速率更快。主要原因是:石墨烯中存在一定的活性缺陷位点,可将吸附H2O 转化为羟基自由基(·OH),攻击破坏甲基橙的共轭发色集团,达到脱色降解效果[27]。但硫掺杂带来的电荷分布改变,可引起碳基体产生应变和缺陷,提升材料的氧化还原性能[33−34]。由元素分析仪与XPS 测试结果可知SGN 的硫原子掺杂量达到10%以上,有研究表明当被掺杂物中的硫原子含量高于4%时,材料会呈现良好的金属特性[35]。因此,高硫原子掺杂量可能提升了SGN的电化学性能[36];与此同时,高含量的硫所带来的C—S 和C—SOx—C 结构能够增加更多新的活性位点[34,37]。所以,硫掺杂可在原有基础上,有效提升材料的氧化还原性能,弥补SGN 缺陷程度低的问题,使其前期降解速率更快。同样图12(b)为初始pH=7、外加电流为20mA、电解质为NaCl、实验温度为20℃条件下,使用RGN和SGN对RBK5溶液(10mg/L)进行降解,可以看出SGN 的降解效率明显高于RGN。综合图12(a)和图12(b)两种染料降解效果的对比,说明硫掺杂对石墨烯性能有一定的提升。
图12(c)为初始pH为7、外加电流为20mA、电解质为NaCl、实验温度为20℃条件下SGN 降解RBK5、罗丹明B 和亚甲基蓝染料,降解率分别达到94.98%、97.83%和76.75%,表明SGN 对其他类型的染料同样具有优良的电催化降解效果。甲基橙与RBK5、罗丹明B、亚甲基蓝分别是单偶氮结构、氧杂蒽型三芳甲烷结构以及杂环偶氮结构[28,31,38−39]。与杂环偶氮结构相比,可能是由于单偶氮结构和三芳甲烷结构化合物的发色基团更易被破坏,所以相同时间条件下亚甲基蓝的降解率明显低于其他染料[40−41]。图12(d)为初始pH为7、外加电流为30mA、电解质为NaCl、实验温度为20℃条件下SGN对4种染料的降解效果。通过图12(c)和图12(d)对比可以看出,将外加电流增大至30mA后,SGN对其他染料的降解效率同样得到明显的提升。由于三芳甲烷结构化合物的发色基团与单偶氮结构相比更易被破坏[41],因此罗丹明B的降解效果提升更加明显。
3 结论
本研究以氧化石墨烯和二苯二硫醚为原料,采用改进高温热退火法成功合成硫掺杂石墨烯。制备硫掺杂石墨烯材料硫元素含量可达到10%以上,并以C—S 和C—SOx—C 结构存在。以染料为目标污染物进行催化效率测试显示,硫掺杂石墨烯对多种染料均有良好的催化性能。电解质种类、外加电流、染料浓度以及初始pH 对甲基橙降解效率均有一定程度的影响,其中电解质作为活性物种产生的影响因素,对材料性能影响最显著。在初始pH为3、甲基橙浓度为10mg/L、电解质为NaCl、外加电流为30mA且实验温度为20℃的最佳条件下,硫掺杂石墨烯电催化降解35min 可以达到98.39%甲基橙的降解率。重复实验显示,硫掺杂石墨烯的稳定性良好,可重复使用。
