泛素-蛋白酶体系统在家族性心肌病发生发展中的作用进展
2021-01-15朱奕潼曹明强
朱奕潼 曹明强
215009 苏州卫生职业技术学院苏州检验医学生物技术重点实验室(朱奕潼);215006 苏州大学附属第一医院心血管内科(曹明强)
在哺乳动物细胞中,大多数蛋白质处于动态流动状态。每个细胞中蛋白质合成和降解的平衡受到高度调控,并以特定的方式发生以维持细胞内稳态。然而,在心脏重构的情况下,这种平衡被打破,导致毒性蛋白质蓄积。为确保错误折叠或异常蓄积的蛋白质得到及时修复或清除,需要一套分子机制单独或协作进行质量控制,包括分子伴侣和共伴侣、自噬-溶酶体通路和泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)[1]。近年来,UPS的变化在家族性心肌病中的作用越来越受到认可。心肌病指的是一组以心脏结构和功能异常为特征的疾病,且排除冠心病、高血压、瓣膜病和先天性心脏病等继发性改变,主要分为肥厚型、扩张型、限制性和致心律失常性右心室心肌病[2]。最常见的家族遗传性心肌病为肥厚型心肌病(hypertrophic cardiomyopathy,HCM)和扩张型心肌病(dilated cardiomyopathy,DCM),通常与编码肌节、细胞骨架、肌浆网、t小管等蛋白的基因突变有关[3]。本文对UPS进行文献综述,在HCM和DCM的背景下,讨论UPS参与的发病机制并提出潜在的干预靶点。
1 UPS简介
UPS是大多数细胞内胞浆蛋白、核蛋白和肌原纤维蛋白高选择性降解不可缺少的部分,控制许多生理过程,如细胞周期、细胞分裂、免疫反应、抗原呈递、细胞凋亡和细胞信号传导等。UPS不仅可以降解底物,而且在转录后水平调控基因的转录活性[4]。
UPS对蛋白质的降解作用是一个ATP依赖的多阶段过程,降解前通过26S蛋白酶体对目标蛋白进行首次泛素化。目标蛋白的泛素化是一个涉及E1(泛素激活)、E2(泛素偶联)和E3(泛素连接酶)在内的酶促级联反应[5]。哺乳动物目前已知有2种E1、40种E2和600多种E3酶。泛素化过程的发生具有空间、时间和底物特异性,这取决于E3泛素连接酶。泛素化过程中,泛素C端羧基通过异肽键与赖氨酸残基胺共价结合。根据泛素分子中7个可用赖氨酸残基的使用情况,形成不同连接方式的泛素链,如K6、K11、K27、K29、K33、K48和K63。泛素链分为线性泛素化、单泛素化、多泛素化、支链泛素化或混合泛素链,不同的链被不同的蛋白质识别并靶向不同的信号通路[6]。
UPS功能的评估方法有很多,使用Western Blot方法可测定泛素化蛋白质的稳定水平,泛素化和去泛素化酶,以及蛋白酶体亚基。此外,荧光合成基质法常用来测定蛋白质的水解活性,缺点是这些小的基质很容易以非泛素化的方式进入20S的核心。为了深入观察进入活体细胞中UPS的动态行为可采用荧光标记UPS组件,包括荧光泛素和荧光标记的蛋白酶体亚基[7]。目前,表达UPS荧光底物的转基因小鼠模型已经建立用以破译UPS在整个动物中的作用[8-9]。
2 UPS与家族性原发性心肌病
UPS功能的损伤足以引起一系列心脏疾病,包括心力衰竭、心肌疾病、肥厚、萎缩、缺血再灌注和动脉粥样硬化,究其原因,至少部分是由于泛素化蛋白积累、蛋白异常聚集(如前淀粉样低聚体形成)以及蛋白酶体活性改变所致[10-11]。目前已有研究发现,家族性HCM和DCM中UPS发生了显著的变化。
2.1 UPS与HCM
HCM是最常见的心脏遗传性疾病,呈常染色体显性遗传,60%~70%为家族性,30%~40%为散发性,家族性病例和散发病例、儿童病例具有同样的致病基因突变[12]。目前已证实,至少14种基因突变与HCM的发病有关,其中有10种是编码肌小节结构蛋白的基因,最常见的突变基因是编码心肌肌凝结合蛋白C(cMyBP-C)的MYBPC3[13]。人类MYBPC3基因突变导致的cMyBP-C破坏后性质不稳定,不能很好地与肌节结合[14]。在纯合子MYBPC3靶向敲入和敲除小鼠发生左心室肥大伴收缩和舒张功能障碍的同时,检测到其蛋白酶体活性升高,且在出生后的前3个月,与左心室肥大的程度呈正相关[15]。除了MYBPC3,MuRF E3连接酶的作用也越来越受到重视。MuRF1是MuRF E3连接酶的成员之一,已被证实可以维持肌细胞内肥大和反肥大信号的平衡,这种平衡是通过抑制肌细胞中蛋白激酶C(protein kinase C,PKC)介导的信号来实现的。PKCs是心肌肥厚的重要触发器,PKCε的激活主要通过Gαq过表达或与活化的蛋白激酶C受体1结合两种方式,最终导致心肌肥厚[16-17]。MuRF1还调节三碘甲状腺原氨酸,这是一种活跃的甲状腺激素,MuRF1可抑制三碘甲状腺原氨酸诱导的生理性心肌肥厚以维持心脏稳态[18]。TnT突变约占HCM病例的7%,Gilda等[19]发现3月龄的严重TnT相关HCM小鼠的心脏UPS功能受损,在这些转基因小鼠模型上进行的蛋白组学研究表明,多个蛋白酶体亚基的表达受到影响。其他致病基因的突变较为罕见,仅在孤立的HCM中被发现,例如编码锚蛋白重复域1的FHL1发生突变的错义和截断表达以及ANKRD1的错义突变,ANKRD1与MuRF1和MuRF2相互作用,提示其可通过MuRF1介导降解[20]。
2.2 UPS与DCM
DCM的特点是心室增大、收缩功能障碍和心肌纤维化[21]。20%~50%的DCM是常染色体显性遗传的家族性疾病[3]。与HCM相比,DCM的基因基础更加多样化,与其相关的单基因大约有50多种[22]。研究发现,DCM心脏中蛋白酶体受损以及泛素化途径发生改变,泛素化途径中不同蛋白的丢失或突变与DCM的发病有关[23]。DCM主要是编码α-B-crystallin的CRYAB和编码desmin 的DES突变引起的。CRYAB或DES突变导致异常蛋白积累,在desmin相关性心肌病(desmin-related myopathy,DRM)中发生严重的DCM[24]。通过CRYAB和Des-D7突变获得的DRM转基因小鼠模型在心力衰竭前便出现了UPS系统的损伤,可能与泛素化蛋白进入20S蛋白酶体的传递相关[25-26]。Murata等[27]发现,小鼠幼年期心脏PRMT1缺失可诱导DCM,并在PRMT1-cKO小鼠中鉴定出从选择性剪接mRNA转译而来的Eif4a2蛋白亚型可被UPS差异泛素化和降解。Li等[28]发现,血浆LMO7基因水平与左室舒张末径、左室舒张末容积、脑钠肽呈负相关,提示LMO7基因可能在汉族人群DCM发病机制中发挥重要作用。DCM患者心肌组织的E3连接酶MAFbx蛋白表达量明显下降[29],Watanabe等[30]发现,心脏E3连接酶FBXO32(Atrogin 1/MAFbx)纯合子突变(Gly243Arg)与DCM有一定的相关性。与DCM相关的其他基因也受UPS介导监管,如编码核纤层蛋白A/C的LMNA,它们是核膜的蛋白质,LMNA突变易诱发DCM,出现传导和(或)节律缺陷[31]。
3 UPS损害的机制和对心肌的影响
基因突变导致UPS受损的机制尚未完全阐明。错误折叠突变蛋白逃脱了伴侣和UPS的监视容易形成聚集物,对细胞有潜在的毒性。Robins等[32]研究表明,人类HCM或DCM心肌细胞普遍发生淀粉样变性。而UPS工作不足本身可能会刺激细胞从适应性反应向促凋亡反应转变。值得注意的是,累加和(或)错误折叠蛋白的聚集本身可能导致UPS受损,从而形成一个有害的反馈回路,造成恶性循环(图1)。
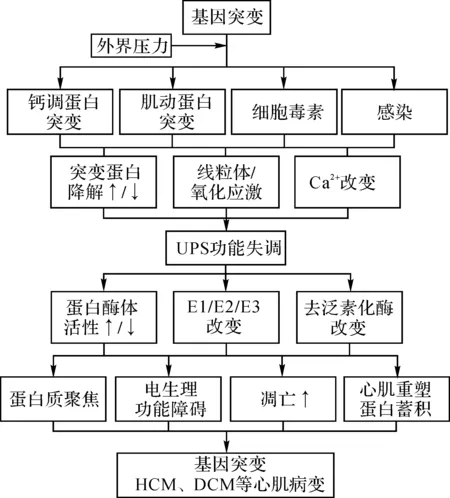
Ca2+:钙离子;UPS:泛素-蛋白酶体系统;E1/E2/E3:泛素激活/泛素偶联/泛素连接酶;HCM:肥厚型心肌病;DCM:扩张型心肌病
UPS损伤可能会对心脏造成多种后果,介导心肌纤维生成和分化的几个信号蛋白,如β-catenin和calcineurin通常由UPS降解[34]。同时,p53是E3泛素连接酶MDM2的靶点[35]。因此,蛋白酶体损伤可引起促肥大和促凋亡因子水平的升高。研究表明,UPS损伤可激活钙调神经磷酸酶-NFAT通路,促进心肌细胞非适应性重构[36]。HCM患者心肌细胞PGlu334LysMYBPC3突变的表达引起UPS损伤,造成促凋亡因子的蓄积和钙离子活动的改变,可导致心律失常[37]。
4 针对心肌病的UPS干预措施
UPS在维持细胞内稳态方面具有中心地位。由于其参与多种重要的调控过程,许多参与泛素化、去泛素化或泛素介导的蛋白质降解被认为是潜在的药物靶点。近年来,蛋白酶体的抑制成为心脏疾病治疗的热点。不可逆蛋白酶体抑制剂环氧米星已被证实可以完全阻止大鼠主动脉横向收缩(transverse contraction of aorta,TAC)[38]。此外,在TAC发生2周后即明显肥大阶段给予环氧酶素,可引起小鼠心肌肥大消退,稳定心功能[39]。相比之下,使用不可逆蛋白酶体抑制剂PS-519治疗小鼠异丙肾上腺素诱导的心肌肥大则效果不明显。然而,关于蛋白酶体抑制剂的心脏保护作用,存在着相互矛盾的数据。硼替佐米是一种新型的二肽硼酸,是第一个进入临床试验的蛋白酶体抑制剂,用于治疗多发性骨髓瘤患者。长期服用硼替佐米诱导左心室肥厚的程度与TAC相似,可导致小鼠心力衰竭和早亡[36]。长期使用可逆抑制剂MLN-273(硼替佐米的类似物)进行治疗可导致模型猪的左心室肥厚、舒张功能障碍和心排血量下降。重要的是,包括老年患者或已存在心脏问题的患者使用硼替佐米后会出现心功能障碍甚至心力衰竭等并发症。故在UPS功能失调的情况下,持续的蛋白酶体抑制剂预计会恶化病情,部分或短期的蛋白酶体抑制剂可能具有心脏保护作用。
另一方面,鉴于在人类和心肌疾病实验模型中观察到的蛋白酶体功能的整体降低,蛋白酶体功能的重新激活被认为是更合适和有益的。Li等研究表明,蛋白酶体增强诱导的超表达蛋白酶体活化剂28α(PA28α)可减少心脏肥大,推迟过早死亡,防止DRM小鼠模型的急性心肌缺血/再灌注损伤,但目前尚无药物可以模拟这种效果。另一种增强蛋白酶体功能的方法是靶向蛋白激酶G。蛋白激酶G可被PDE5抑制剂西地那非激活以提高cGMP水平。西地那非在心力衰竭患者和动物模型中可诱导逆转重构和改善左室舒张功能,可能是一种新的治疗方法。
此外,泛素化过程可通过去泛素化酶(DUBs)逆转。Liu等[41]研究发现,泛素特异性蛋白酶14去泛素酶活性降低导致心肌肥厚特异性标志物b-MHC表达水平降低,进而导致GSK-3b磷酸化水平降低,表明泛素特异性蛋白酶14可能是心肌肥厚治疗的新靶点。
5 小结
UPS功能不全的病因及其引起心肌疾病的机制尚处于探究的起步阶段,尚需对UPS在心肌疾病中的病因和作用进行深入研究。对于家族遗传性心肌病的治疗而言,蛋白酶体的整体抑制可能是有害的,蛋白酶体功能的重新激活是一个更有前途的治疗方法。今后仍需在心脏的细胞模型、动物模型和人体模型中寻找UPS的特异性靶点,以提高心肌病的疗效。
利益冲突:无
