Gut microbiota mediated molecular events and therapy in liver diseases
2021-01-15XiaoqiangQiMingYangJosephStenbergRahulDeyLeslieFogweMuhammadShawkatAlamEricKimchiKevinStaveleyCarrollGuangfuLi
Xiaoqiang Qi, Ming Yang, Joseph Stenberg, Rahul Dey, Leslie Fogwe, Muhammad Shawkat Alam, Eric T Kimchi, Kevin F Staveley-O'Carroll, Guangfu Li
Abstract Gut microbiota is a community of microorganisms that reside in the gastrointestinal tract. An increasing number of studies has demonstrated that the gutliver axis plays a critical role in liver homeostasis. Dysbiosis of gut microbiota can cause liver diseases, including nonalcoholic fatty liver disease and alcoholic liver disease. Preclinical and clinical investigations have substantiated that the metabolites and other molecules derived from gut microbiota and diet interaction function as mediators to cause liver fibrosis, cirrhosis, and final cancer. This effect has been demonstrated to be associated with dysregulation of intrahepatic immunity and liver metabolism. Targeting these findings have led to the development of novel preventive and therapeutic strategies. Here, we review the cellular and molecular mechanisms underlying gut microbiota-mediated impact on liver disease. We also summarize the advancement of gut microbiota-based therapeutic strategies in the control of liver diseases.
Key Words: Gut microbiota; Intrahepatic immunity; Metabolite; Fecal microbial transplantation; Probiotic; Antibiotic
INTRODUCTION
The growing evidence of gut microbial roles in human diseases attracts researchers’ attention in exploring gut microbiota-mediated therapy. The gut microbiota is defined as the entire community of microorganisms residing in the gastrointestinal tract, and it is dominated mainly by bacteria[1]. In the earlier stages of investigation, people focused solely on the gut microbiota’s function regarding modulation of human nutrition and metabolism, on which a healthy status relies. However, dysbiosis of the gut microbiome may cause inflammatory bowel disease and bacterial infection in addition to impairing human alimentation[2]. Nowadays, growing evidence has emerged to demonstrate how gut microbiota profoundly and systemically influences human health and disease through various mechanisms, particularly influencing cancer. It has been discovered that gut microbiota and their associated metabolites are closely related to malignant tumor generation by developing chronic inflammation and immune surveillance dysregulation.
More interestingly, due to the close anatomical and physiological connection between the gut and liver, the role of gut microbiota and its associated metabolites were found in liver diseases from many studies (discussed in the following sections). By focusing our attention on the interactions between gut microbiota and liver diseases, we are pulling back the curtain on how gut microbiota and its associated metabolomes influence liver health and disease. However, it seems as though these findings are but the tip of the iceberg. Many questions still remain. Some cause-specific and disease severity-specific microbiota in liver diseases have been reported, but the underlying mechanisms are still unclear or only show a piece of the whole picture. For example, there are distinct explanations for antibiotics cocktail (ABX) mediated suppression of hepatocellular carcinoma (HCC) tumor progression. Therefore, it is not surprising that many different independent studies targeted the same question to attain different answers, as gut-mediated effect on the liver is involved by a wide range of factors. This further confirms that interactions amongst the gut microbiota and liver work through multiple pathways. These interactions may have developed over a long period as a result of evolution. A systemic review of recent research findings in the gut-liver axis helps to dig the underlying mechanism of how the gut modulates liver function and disease treatment.
Here, we discuss the current findings in correlations between gut microbiota and categorized liver diseases. The perspectives of underlying mechanisms will be reviewed and summarized. Development in gut-microbiota-manipulation-based preventive and therapeutic strategies in liver diseases also will be considered. Given all the current progress, gaps in understanding the influence of gut microbiota on liver diseases are proposed, and the feasible directions are prospectively assumed.
DYSBIOSIS OF GUT MICROBIOTA FACILITATES LIVER DISEASES
Nonalcoholic fatty liver disease and alcoholic liver disease
Nonalcoholic fatty liver disease (NAFLD) and the advanced stage nonalcoholic steatohepatitis (NASH) are currently the most common types of liver disease in humans. Studies have indicated that alteration of gut microbiota is involved in the development of NAFLD and NASH[3,4]. Endotoxemia induced by increased gut permeability has been observed in patients with NAFLD, suggesting that gut permeability-induced inflammatory pathways contribute to NAFLD pathogenesis[5]. Small intestinal bacterial overgrowth (SIBO) is another often observed dysbiosis of gut microbiota occurring in NAFLD/NASH[3,6]. Along with the altered intestinal microbiota profiles, SIBO was seen in most patients with cirrhosis[7]. The presence of SIBOs may partially explain why the permeability of the gut increases. Gut permeability and SIBOs enhance the hepatic expression of Toll-like receptor 4 (TLR-4) and the production of interleukin 8 (IL-8). Therefore, determining the specific pathogenic bacteria species has been made a very appealing question. Clinical studies revealed that patients with NASH exhibited significantly lower concentrations ofBacteroidetesin their gut compared to healthy individuals[4,8]. Meanwhile, another cohort study showed that high-alcohol-producingKlebsiella pneumoniaewas associated with up to 60% of NAFLD patients[9]. Selective depletion ofK. pneumoniaebefore fecal microbiota transplant (FMT) from NASH patients into mice prevented NAFLD development in recipient mice. In contrast, the study on patients with alcohol-induced liver cirrhosis exhibits a decreased proportion ofBacteroidaceaefamily than healthy individuals[10]. Also, the analysis of the gut microbiota profiles in patients with cirrhosis revealed an increase in pathogenic bacteria and a decrease in beneficial bacteria, such as a decreasedBacteroidetesalong with overgrowth of theProteobacteriaandEnterobacteriaceaespecies[11,12]. However, it remains unclear if this change drives disease or it comes from disease as a result. To answer this question, a well-designed, largescale clinical and experimental investigation is needed. Moreover, in excessive alcohol intake induced gut microbiota dysbiosis, the intestinal innate immune responses, such as secreted antimicrobial molecules, may impact disease development. For example, the reduced bactericidal c-type lectins, Reg3b, and Reg3g in the small intestines were discovered after alcohol feeding, further causing SIBO and dysbiosis in mice[13].
HCC
HCC, the final stage of chronic liver disease, has been evidenced to have gut microbiota involved in its initiation and progression. In DEN/CCl4induced HCC murine model, germ-free mice presented fewer and smaller tumors compared to its syngeneic parallel[14], indicating that gut commensal flora is required for tumor progression and the activation of TLR-4 by lipopolysaccharide (LPS) promotes tumor generation. In HCC murine models, studies also demonstrated that the administration of ABX in tumor-bearing mice effectively slowed tumor progression[15,16]. Even though the underlying mechanisms were distinct in those studies, which will be further discussed in the following sections, we can assume modulating gut microbiota is favorable to prevent liver cancer. However, there is still a lack of clinical data to support it. In addition, obesity-induced gut microbiota dysbiosis that produces DNA damaging bacterial metabolites has been indicated to facilitate HCC development in high-fat diet (HFD)/carcinogen 7,12-dimethylbenz[a]anthracene treated murine model. Furthermore, the administration of antibiotics was able to reduce the prevalence of tumors in the chemically treated mice[17]. However, in those studies, the relevant specific bacteria species remain unclear. Clearly identifying either specific pathogenic or beneficial bacteria species may be the next mission.
MODULATION OF INTRAHEPATIC IMMUNITY, THE CENTRAL ROLE OF GUT MICROBIOTA IN LIVER DISEASE
Although etiologies vary in liver diseases, inflammation and liver fibrosis are the basis for most liver diseases. The intrahepatic immunity has been certified as an acting point, as gut microbiota can play different roles in liver inflammation and establishment of fibrosis through different liver infiltrating immune cells and resident hepatic cells[18]. Furthermore, the immune checkpoints emerged as a central pivot to modulate disease progression with gut microbiota-derived metabolites as informatic messengers (Figure 1) and various TLRs as informatic receivers.
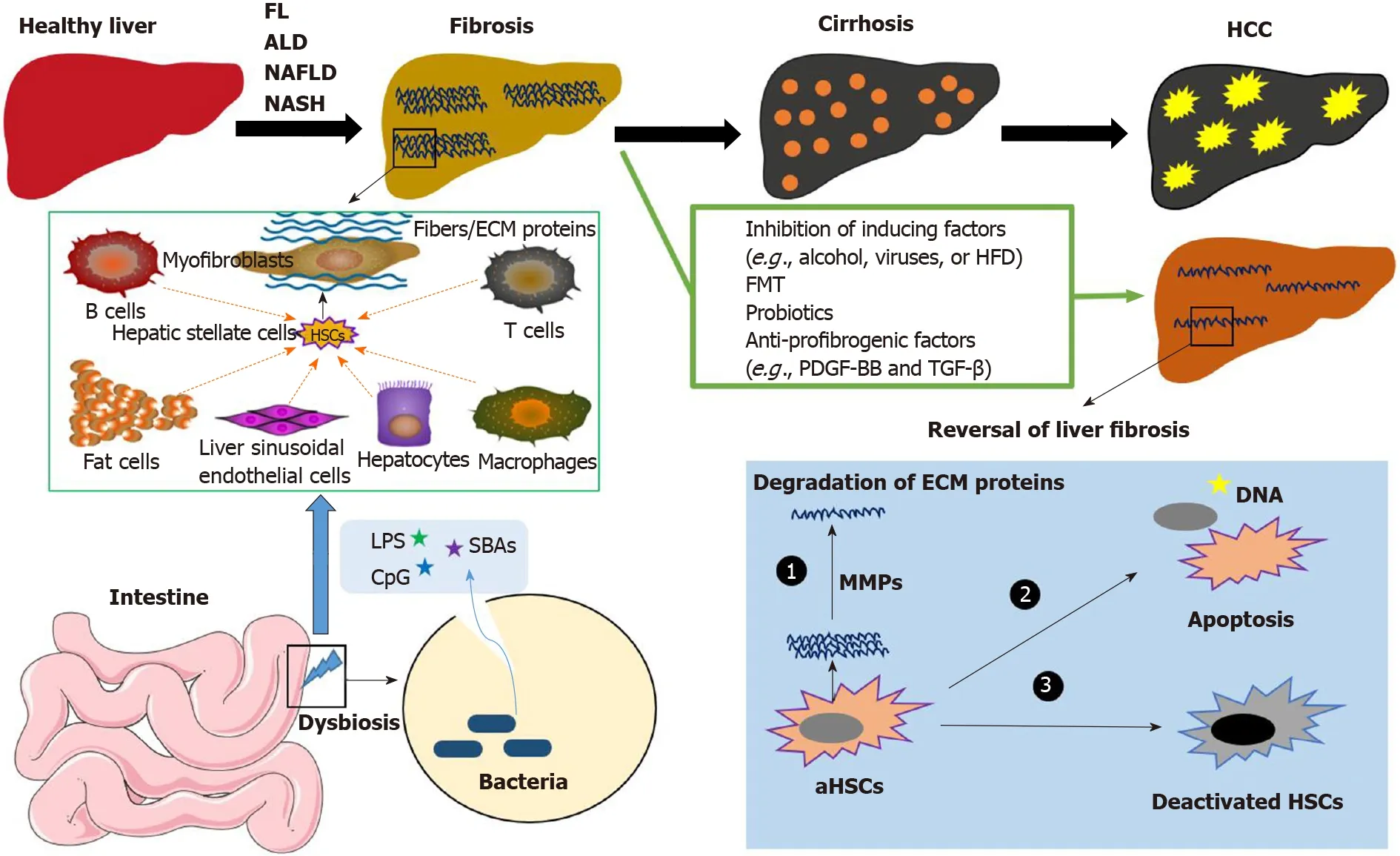
Figure 1 The development of liver diseases. Without effective treatment or preventive strategies, fatty liver disease, alcohol-induced liver disease, nonalcoholic fatty liver disease, and nonalcoholic steatohepatitis can result in liver fibrosis, cirrhosis, and hepatocellular carcinoma. Gut microbiota-derived molecules, including lipopolysaccharide, CpG, and secondary bile acids, initially activate liver resident cells to produce cytokines and chemokines. Quiescent hepatic stellate cells (HSCs) can be activated and transformed into myofibroblasts (MFBs), which is mediated by chemokines and cytokines released by liver-infiltrating macrophages, leukocytes, and other cell types, including fat cells and damaged hepatocytes. MFBs are the predominant source of collagen-producing cells and other extracellular matrix proteins (ECM). With effective treatment, such as fecal microbiota transplant, probiotics, and anti-profibrogenic factors, fibrosis is reversible. The treatments that induce apoptosis (or deactivation) of activated HSCs or MFBs and degrade the ECM proteins can reduce the stiffness of the liver, reverse liver fibrosis, and inhibit the progression of liver disease. FL: Fatty liver; ALD: Alcohol-induced liver disease; NAFLD: Nonalcoholic fatty liver disease; NASH: Nonalcoholic steatohepatitis; HCC: Hepatocellular carcinoma; ECM: Extracellular matrix proteins; HFD: High-fat diet; FMT: Fecal microbiota transplant; LPS: Lipopolysaccharide; SBAs: Secondary bile acids; HSC: Hepatic stellate cells.
Hepatic resident and infiltrating cells
Hepatic cells are comprised of parenchymal hepatocytes and the non-parenchymal cells (NPCs), including quiescent hepatic stellate cells (HSCs), Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs), and diverse lymphocytes (e.g., natural killer cells, NKT cells), which play crucial roles in the liver health and disease[19,20]. Here, we discuss how gut microbiota modulates intrahepatic immune responses by different hepatic cell types.
Hepatocytes:Hepatocytes consist of two-thirds of the total liver cell population and are responsible for the primary metabolic functions and engage in the immune responses by interacting with NPCs[21]. In different mouse models of liver fibrosis, either induced with alcohol or non-alcohol (e.g., CCl4or HFD), injured hepatocytes release reactive oxygen species and fibrogenic mediators, which recruit lymphocytes and promote the activation of HSCs[22]. The Notch signaling pathway also plays a pivotal role in liver development, liver repair, and carcinogenesis[23]. Notch activation in hepatocytes can induce and promote liver fibrosis in normal chow diet and NASH diet-fed mice[24], respectively. Even though the mechanism by which the gut microbiota directly impacts the Notch signaling in hepatocytes is unclear, some studies have shown that dysbiosis of gut microbiota may increase the activation of the Notch signaling pathway, thus promoting tumor regression[25]. In addition, the TLR signaling pathway of hepatocytes is directly involved in the gut-liver axis, which will be discussed in the following context.
HSCs:HSCs reside in the space of Disse (perisinusoidal space) between the hepatocytes and LSECs. Their main functions include the storage of vitamin A and lipid droplets[26]. HSCs are the primary sources of liver myofibroblasts[27]and play a key role in the initiation, progression, and regression of liver fibrosis[28]. All other liver cells can directly or indirectly impact the activation, trans-differentiation of HSCs to myofibroblasts, and their deactivation. There are multiple cellular and molecular signals involved in these reactions[29]. The interaction of HSCs with other hepatic cells is bidirectional. For instance, upon activation, HSCs, in turn, can further activate macrophages by secreting macrophage colony-stimulating factor (M-SCF), MCP-1, IL-6, RANTES, and so on[30].
KCs:Liver macrophages, including the liver resident KCs and circulating monocytederived macrophages, play essential roles in various liver diseases, including virus hepatitis, alcoholic liver disease (ALD), NAFLD, NASH, and HCC[31,32]. The purpose of macrophages in NAFLD and NASH and the underlying mechanisms involved in liver inflammation and liver fibrosis have been well summarized recently[33]. Gut-derived endotoxins (LPS), lipid metabolites, and hepatocyte damage-associated molecules are the main factors inducing macrophage activation in NAFLD[33]. Furthermore, the activated macrophages secrete chemokines (e.g., CCL2) and cytokines (e.g., TGF-β1), which leads to the activation of T cells and the transformation of HSCs into myofibroblasts.
LSECs:With the loss of fenestration, capillarized LSECs reduce the transfer of nutrients and other products from the blood to hepatic cells[34,35], which occurs with liver fibrosis in animals and humans[36]. The capillarized LSECs can secrete TGF-β1 and contribute to the accumulation of extracellular matrix proteins, including fibronectin and laminin in the liver, promoting the activation of HSCs and contributing to the formation of liver fibrosis[35]. Xieet al[37]reported that the reversal of LSEC differentiation with the use of BYY 60-2770, an activator of soluble guanylate cyclase (sGC), prevented the progress of rat liver cirrhosis. LSECs are directly exposed to dietary and bacterial products from the gut through the portal circulation. It has been shown that there is a link between the fenestration of LSECs and diet-induced changes in the gut microbiome. The increase of fenestration of LSECs was associated with a high abundance ofFirmicutesand a low amount ofBacteroidetes[38].
NKT cells:In vivoanimal studies showed that alteration of gut bacteria with ABX treatment induced a liver-selective anti-tumor effect with an increase of hepatic CXCR6+NKT cells and heightened IFN-γ production upon antigen stimulation[15]. NKT cell accumulation was regulated by CXCL16 expression of LSECs, which is controlled by gut microbiome-mediated primary-to-secondary bile acid (SBAs) conversion[15]. Similarly, Zhanget al[39]reported a significant increase of primary bile acids (PBAs) in the large intestine of mice treated with antibiotics such as vancomycin and imipenem. This antibiotic treatment was also associated with the selective suppression of several bacterial species such asClostridium,responsible for deconjugating PBAs into SBAs. Selective depletion of intestinal bacteriaFirmicutesincreases PBA levels, which further promotes hepatic NKT cell accumulation and NKT-mediated anti-tumor effect, indicating that modulating gut microbiota is an optional strategy for HCC treatment.
Immune checkpoints
The efficacy of anti-immune checkpoints (PD-1 or CTLA4) in cancer treatment was markedly reduced in germ-free mice or specific-pathogen-free mice treated with broad-spectrum antibiotics[40]. For instance, the efficacy of anti-CTLA4 on melanoma immunotherapy in mice and patients was associated withBacteroides fragilis, since gavage withB. fragilisor adoptive transfer ofB. fragilis-specific T cells recovered the anti-tumor efficacy of CTLA4 blockade in antibiotic-treated or GM mice[41]. Similarly, the abundance ofAkkermansia muciniphilawas associated with the effectiveness of anti-PD-1 immunotherapy[42]. FMT from cancer patients who were not responding to anti-PD-1 treatment to tumor-bearing mice failed to ameliorate the anti-tumor effects of anti-PD-1. In contrast, oral supplementation ofA. muciniphilarestored the anti-tumor efficacy of anti-PD-1 in those mice[42]. Meanwhile, Gopalakrishnanet al[43]also reported the gut microbiota modulated therapeutic responses to anti-PD-1 immunotherapy in human melanoma patients. Iidaet al[44]concluded that antibiotic-induced disruption of gut microbiota impaired both CpG oligonucleotide immunotherapy and platinum chemotherapy in their subcutaneous breast cancer murine model. Investigation on anti-immune checkpoints on HCC has recently begun[45,46]; however, the role of gut microbial effects on the blockage of immune checkpoints in liver diseases remains unclear. Notably, immune checkpoint inhibitors-associated hepatotoxicity should not be ignored[47,48]. For example, clinical trials have shown that alanine aminotransferase elevations in grade 3-4 levels are observed in approximately 20% of patients who are treated with ipilimumab (CTLA-4 inhibitor) and nivolumab (PD-1/PD-L1 inhibitor)[49].
Metabolites
Currently, the role of microbiota in liver diseases has been broadly investigated[50]. Gut microbiota-associated metabolites such as fatty acids, amino acids, and carbohydrates are a group of contributory factors involved in the modulation of intrahepatic immune responses[51]. Maet al[15]observed that manipulation of gut microbiota by treatment with an ABX can suppress liver tumor growthviaaccumulated NKT cells, which increased anti-tumor immunity in the liver. Modulating commensal gut microbiota composition with ABX treatment changed the bile acid metabolism and significantly increased the PBA production in the liver microenvironment. This rise in PBA production elevated CXCL16 expression on LSECs, which recruited more CXCR6 specific NKT cells to the liver. Conversely, SBAs have been shown to reverse this phenomenon and reduce CXCL16 expression on LSECs. As such, alteration of the ratio of PBAs to SBAs resulted in the growth of hepatic NKT cell accumulation in the ABX treated mice compared to control mice. In addition, the conversion of PBAs to SBAs can be significantly inhibited with the treatment of antibiotics. SBAs, such as deoxycholic acid, activated mTOR to promote the HCC development in NASH mice fed with a steatohepatitis-inducing high-fat diet (STHD-01)[52]. Accumulation of free fatty acids in hepatocytes can also induce lipotoxicity, which causes hepatocellular injury and NAFLD progression[53]. Similarly, excess carbohydrates from dietary intake can bede novosynthesized to lipids in the liver, causing lipotoxic liver injury[54]. The characteristics of metabolites of gut bacteria with the association of liver diseases found in recent studies are summarized in Table 1. The mechanisms of how microbiota-derived metabolites might influence liver diseases or their potential roles as diagnostic markers have been summarized when we were drafting this review[55-61].
TLRs
The TLR family is one of the best-characterized families of pattern recognition receptors[62], which can activate the innate immune system by recognizing pathogenassociated molecular patterns and damage-associated molecular patterns[63]. There are a total of 13 TLRs in mammals; only TLR1 to 10 exist in humans[64]. TLRs, excluding TLR10, can bind with different bacteria-derived molecules to participate in the inflammatory and immune responses[65]. The gut-liver axis defines the close anatomic connection, through the circulation of the portal vein and biliary tract, and the functional interaction of the gastrointestinal tract and the liver[66]. Through the portal vein, bacteria-derived products, including LPS and CpG-containing DNA, might regularly activate TLR signaling pathway in the hepatocytes and other hepatic cells. Current studies have demonstrated that TLR signaling pathways play pivotal roles in liver inflammation, fibrosis, regeneration, and carcinogenesis[67,68]. TLRs are broadly expressed in hepatic cell populations, including hepatocytes, LSECs, KCs, lymphocytes, DCs, biliary epithelial cells, and HSCs[68], which link the gut-liver axis. In this review, we summarize the recent findings of the role of TLRs in liver diseases.
TLR1, TLR6, and TLR10:Currently, TLR10 is the only member of the human TLR family without a definite known ligand, function, and localization[69]. Phylogenetic analysis shows that TLR10 is most related to TLR1 and TLR6, both of which mediate immune responses in cooperation with TLR2[70]. The study of chimeric receptors of TLR10 indicated that it can sense triacylated-lipopeptides and other microbial-derived agonists, the ligands of TLR1[70]. We will not describe the separate roles of these three TLRs at this point.
TLR2:Liver injury resulting from adenovirus or CCl4promoted more robust liver inflammation by releasing the TLR2/TLR4 ligands, including HSP60, gp96, HMGB1[71]. The amplified non-autoimmune hepatitis (AIH)-inflammation could result in the initiation of AIH, a severe autoimmune liver disease, which can lead to fibrosis, cirrhosis, and HCC. Conversely, TLR2 deficiency promoted HCC development in C57BL/6 mice associated with the increase of Ly6ChighIL18Rα+myeloid-derived suppressor cells and with impaired CD8+T cells function[72]. This result further confirmed the previous finding that TLR2 knockdown by siRNAs inhibited the proliferation of HCC cell line BLE-7402, which was associated with a reduction of IL-6 and IL-8 production[73].
TLR3:TLR3 activation by agonist double-stranded RNA BM-06 or poly(I:C) induced apoptosis of HepG2.2.15 HCC cells[74]. The analysis of human HCC tissue also indicated that the expression of TLR3 positively correlated with apoptosis of HCC cells and negatively correlated with the proliferation of HCC cells and angiogenesis[75], suggesting that TLR3 expression may serve as a prognostic marker of HCC.
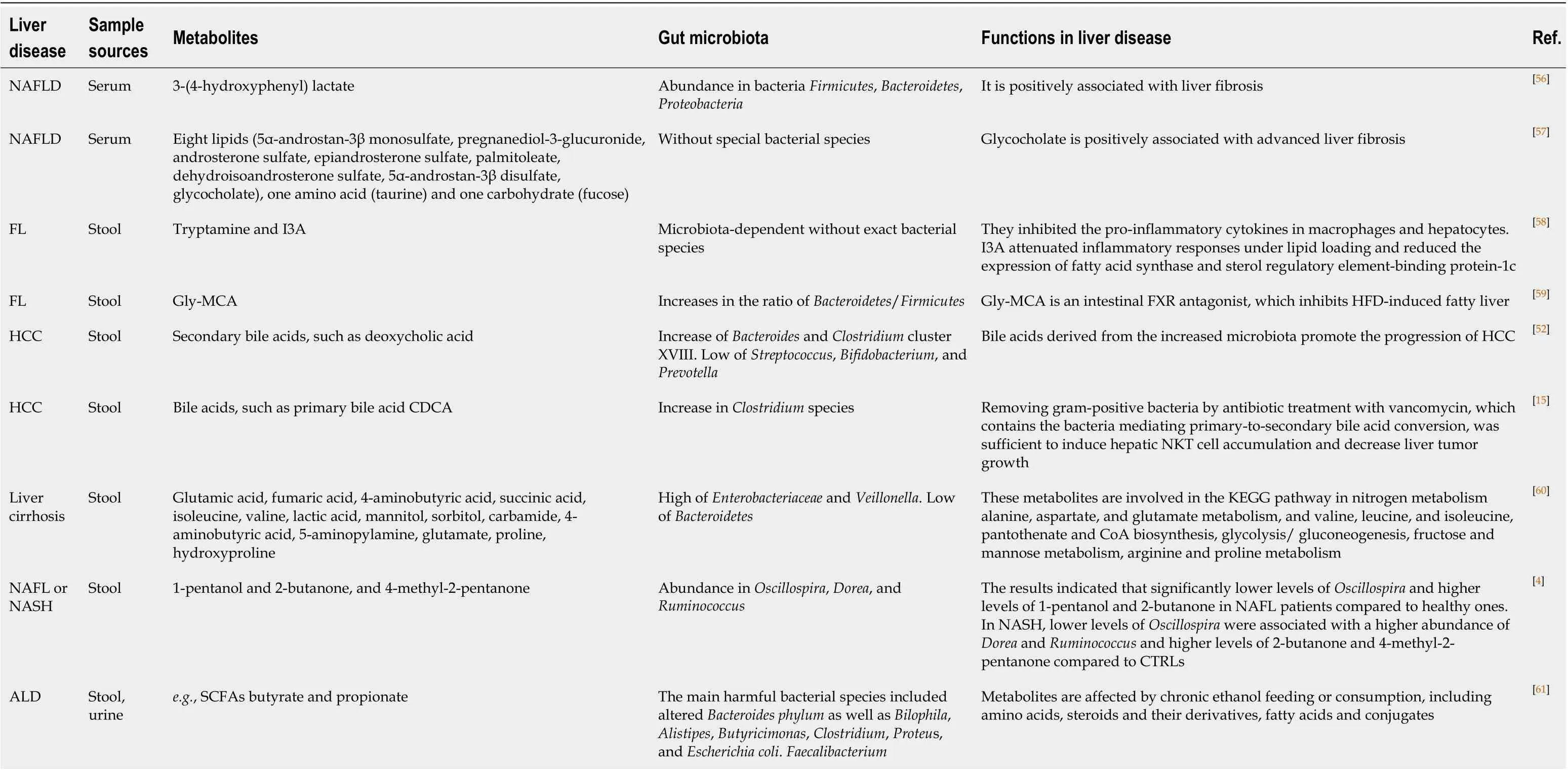
Table 1 Summary of metabolites associated with liver diseases
TLR4:TLR4 is one of the most well-known TLRs implicated in the liver inflammationfibrosis-cancer axis. TLR4 Ligands, including LPS, can induce liver fibrosis by promoting the transformation of HSCs to myofibroblasts through the TLR4-MyD88-NF-κB signaling pathway[76]. Dapitoet al[14]reported that TLR4 was required for HCC progression but not the initiation, which is relative not limited to the increase of hepatomitogen epiregulin. Suppressing the TLR4 signaling pathway in HSCs by Dectin-1, the major progenitors of myofibroblasts, inhibited hepatic fibrosis and hepatocarcinogenesis[77]. Moreover, blockage of TLR4 also potentially inhibited the LPS induced inflammatory reaction and other immune responses[78].
TLR5:TLR5 knockout can attenuate CCl4-induced liver fibrosis in wild-type C57BL/6 mice and the activation of HSCsviainhibiting NF-κB and MAPK signaling pathways[79]. Through the same signaling pathway, TLR5 knockdown also can ameliorate hyperammonemia (HA)-induced liver injury in rats by inhibiting hepatocyte apoptosis, inflammation, and oxidative stress[80].
TLR7:TLR7 activation by its natural ligand imiquimod can induce autophagy and release of insulin-like growth factor (IGF-1) and inhibit lipid accumulation in hepatocytes induced by unsaturated fatty acid (arachidonic acid: oleic acid = 1:1)in vitro[81]. More so,in vivoexperiments also showed that TLR7 knockout prevented NAFLD progressionviainducing autophagy and the release of IGF-1 from the liver.
TLR8:Even though not many studies about the role of TLR8 are reported currently, its role in liver disease should not be ignored. Stimulation of liver intrasinusoidal cells with TLR agonist 1/2, 2, 2/6, 3, 4, 5, 7, 8, or 9 (respectively Pam3CSK4, HKLM, FSL-1, poly(I:C), LPS, flagellin, imiquimod, ssRNA40, or CpG ODN2216) indicated that only the TLR8 agonist ssRNA40 selectively can activate the innate immune cells to produce IFN-γ[82].
TLR9:Tak1ΔHep mice with hepatic deletion of transforming growth factor-β-activated kinase 1 (Tak1ΔHep) can develop a spontaneous liver injury, inflammation, fibrosis, and HCC, mimicking the progression of human HCC[83]. Ablation of TLR9 or TLR4 suppressed the spontaneous process liver inflammation-fibrosis-cancer axis in Tak1ΔHep mice. The inhibition of HCC progression was associated with downstream signaling molecules MyD88 and TNFR.
MANIPULATION OF GUT MICROBIOTA, A PROMISING THERAPEUTIC STRATEGY IN LIVER DISEASE
We have previously highlighted the strong correlations between liver disease and the intestinal microbiota. Further, the modulation of gut microbiota and its products may prove to be a promising target for liver disease and HCC treatment. For example, the abundance ofEnterococcus faecaliswas associated with liver disease severity and the mortality of patients with alcoholic hepatitis due to cytolysin production[84]. Treatment withE. faecalis-targeted bacteriophages can ameliorate alcohol-induced liver disease in humanized mice by reducing liver cytolysin. The intestinal microbiome contains seven-fold more genetic material than the human genome, providing an abundance of potential therapeutic targets. Here, we focus on current studies involving probiotics, FMT, and antibiotics in liver disease treatment.
Probiotics, a new use of an old remedy against liver diseases
Probiotics, prebiotics, and synbiotics have recently undergone investigation for their ability to influence the gut microbiota composition[2]. Research has long established that probiotic yogurt can positively impact gut health[85], and more so, the prebiotic insulin was shown to reduce hepatic lipogenesis and plasma triglyceride concentrations in human patients[86]. Further, the use of probiotics to combat the altered gut microbiota profile in liver disease is currently being investigated in clinical trials[87]. One recent randomized, double-blind, placebo-controlled study investigated the effects of probiotic treatment on intrahepatic fat fractions in 68 patients with NAFLD. They reported that patients exhibited decreased intrahepatic fat fractions after 12 wk of probiotic treatment (from 16.3% ± 15.0% to 14.1% ± 7.7%,P= 0.032), with an overall mean decrease of 2.61% when compared to baseline measurements[88]. Although the argument that this statistical significance may not be clinically significant, the authors propose a mechanism by which specific probiotic agents may aid in correcting dysbiosis rather than directly decreasing hepatic fat. Moreover, this may lead others to investigate the use of probiotics for correcting dysbiosis in liver disease.
FMT, a star of tomorrow to control patient’s gut microbiota in personalized therapy
FMT is also an old yet underrepresented area of investigation. This technique has gained considerable attention after its effectiveness in the last resort treatment of antibiotic-resistant infections such asClostridium difficile[89]. The advantage of FMT from healthy persons to patients is the resetting of gut microbiota composition, returning the flora to a “healthy” state[87]. To investigate the effects of FMT in liver disease, one study used fecal material from patients with ALD to transplant into germ-free mice through oral gavage. The investigators reported increased intestinal permeability and bacterial translocation, resulting in severe liver inflammation in the microbiome repopulated mice[90]. Similarly, a pilot study containing eight male patients with severe alcoholic hepatitis were treated with nasoduodenal FMT over the course of 7 d. Results revealed the resolution of ascites and hepatic encephalopathy (HE) within one week in most FMT treated patients, along with significantly improved 1-year survival rates (87.5%vs33.3%;P= 0.018) compared to historical controls[91]. Taken together, these results demonstrate the importance of the intestinal microbiome population in the development and treatment of liver diseases. While FMT provides distinct challenges for its commercial use in the clinical setting, its profound therapeutic effects can no longer be ignored when current first-line therapies lack adequate efficacy.
Antibiotics, perhaps another resolution in fighting liver diseases
Treatment with antibiotics is the most obvious approach for modulating the intestinal bacterial profile and its products. It has been well documented that microbiota depletion with ABX results in a significant reduction of tumor number, size, and fibrosis severity in several HCC murine models[14,15,17,76]. While ABX treatment is unfit for human studies, antibiotics have undergone investigation to treat liver disease in many clinical trials. The overwhelming majority of current antibiotic studies utilize the antibiotic rifaximin. Further, this section will primarily report current findings related to rifaximin treatment and liver disease; however, ABX and norfloxacin treatments will also be briefly discussed.
Rifaximin:Rifaximin is a highly favorable antibiotic due to its safety profile and broad-spectrum effects against gram-positive and gram-negative aerobic and anaerobic bacteria. Its classification of a non-absorbable antibiotic makes it well suited for targeting the intestinal microbiome rather than treating systemic infections. Unique to most antibiotics, rifaximin works by binding to the beta-subunit of bacterial DNAdependent RNA polymerase, consequently blocking bacterial transcription[92]. Further, this results in endotoxin-lowering and anti-inflammatory effects rather than diminishing the gut flora composition[93-95]. Multiple studies have correlated rifaximin treatment with reduced risk of death and cirrhotic complications in patients with HE[96-98]. More so, a phase 3, open-label maintenance study revealed long-term treatment (≥ 24 mo) of rifaximin (550 mg, twice daily) showed no increased adverse events in HE patients compared to placebo controls, further confirming its safeness of use[98]. However, patients with cirrhosis lack average BA concentrations, resulting in decreased efficacy of rifaximin. To combat this issue, rifaximin soluble solid dispersion (SSD) was introduced. Unlike the previous generation, rifaximin SSD is water-soluble and no longer dependent on the patient’s intestinal BA concentrations. Despite this distinct advantage, few clinical trials have implemented rifaximin SSD into their liver disease studies.
It is widely theorized that liver cancer progression is partly mediated by gut dysbiosis and other downstream effects. Further, we believe a longitudinal study should be performed to assess the preventative effects of rifaximin and rifaximin SSD on liver cancer development in a high-risk patient population. While it is highly unlikely that rifaximin would provide a therapeutic effect in patients who have already developed tumors, the benefits of long-term, preventative rifaximin treatment on patients with a high risk of developing HCC may provide a better response than the current treatment options.
ABX:Besides enhancing anti-tumor immunity in the liver, antibiotics may also weaken the tumor-promoting impact of specific intestinal bacteria. One study investigated how antibiotic alteration of the gut microbiota impacted liver carcinogenesis by depleting the intestinal flora of mice through regular treatment of ABX. Further, results indicated a significant reduction in tumor number, size, and liver-body weight ratio compared to the untreated control mice[14]. More so, the authors observed that antibiotics mitigated tumorigenesis in a healthy liver but did not impact tumors that had already developed. These results suggest that the gut microbiota plays a role in preventing hepatic proliferation and fibrogenesis, leading to liver carcinogenesis. And also, there was a decreased expression of NF-kB regulated genes and increased apoptosis rates in gut-sterilized mice[14], indicating that the gut flora and its products may promote liver inflammation and carcinogenesisviaactivating hepatic TLR-4, further exerting pro-survival signals on hepatocytes.
Norfloxacin:Norfloxacin antibiotics may also attenuate pro-inflammatory phenomena exerted by the immune system. Zapateret al[99]observed that selective intestinal decontamination, facilitated by the antibiotic norfloxacin, in patients with cirrhosis reduced serum and ascitic concentrations of pro-inflammatory cytokines TNF-α, IFNγ, and IL-12 by blunting the activation of serum NF-κB. Furthermore, norfloxacin treated patients exhibited both decreased levels of neutrophilic oxidative burst and apoptotic events. The authors did not describe a precise mechanism by which the fluoroquinolone mitigated immunological processes, though they did propose that norfloxacin exerted a direct effect on cellular function.
CONCLUSION
Here we have described many reported mechanisms that the gut-liver axis plays in liver disease. Further, we aim to spread awareness of the existing knowledge gap that could be used to modulate the gut-liver axis to alter the disease state. The modulation of the intestinal microbiome has transitioned to clinical trials for its use in both preventative and therapeutic approaches in human liver disease. A summary of the current ongoing clinical trials can be found in Table 2. It should be recognized that alteration of the gut microbiota is not a one size fits all solution due to its highly individual nature and convoluted intricacies. However, the bilateral relationship of the gut-liver axis cannot be ignored when its alteration possesses abundant therapeutic potential.
To aid in furthering the use of gut microbiota-based therapy against liver diseases, we will give brief suggestions for future directions in this field. Lacking from clinical data are well-designed, large-scale investigations of the gut flora profile for liver disease progression (e.g., NAFLD, NASH, HCC,etc.). Understanding the underlying mechanisms of how the gut microbiota or its metabolic products alter liver immunity over time could help clarify liver disease development. Such profiles could provide a model to test preventative treatments or therapeutic approaches with antibiotics, probiotics, and FMT. Establishing which bacteria to eliminate and which to nurture would allow for tailored probiotics and specific antibiotics for chronic liver disease therapy. Continuing with this strategy comes the need for longitudinal studies to assess the preventative and therapeutic effects of antibiotics (e.g., rifaximin and rifaximin SSD) on liver disease development. As stated previously, rifaximin SSD would be an ideal drug to study its preventative measures against high-risk liver cancer populations. Pre-, pro-, and synbiotic strategies are underwhelming in clinical studies due to a lack of high-quality study design. Our review highlighted how a decreased level ofBacteroidetesis seen in patients with NASH, cirrhosis, and other liver diseases. Finding commonalities in the abovementioned gut profiles (e.g., decreasedBacteroidetes) will prompt the use of gut microbiome repopulation therapies in clinical trials. Lastly, the use of FMT treatment desperately needs a more substantial safety investigation. Conducting a proper prospective cohort study with a large study size and a longer observation period is crucial for its safe implementation in the clinical setting.
Collectively, modulating the population of the gut microbiome by biological products may be a new generation therapeutic strategy for liver disease. For now, we must wait to see the results of ongoing and scheduled clinical trials. As we wait in anticipation for the results of mentioned clinical trials, we hope that our current knowledge of the gut-liver-axis inspires new ideas for the next potential therapies in liver disease.
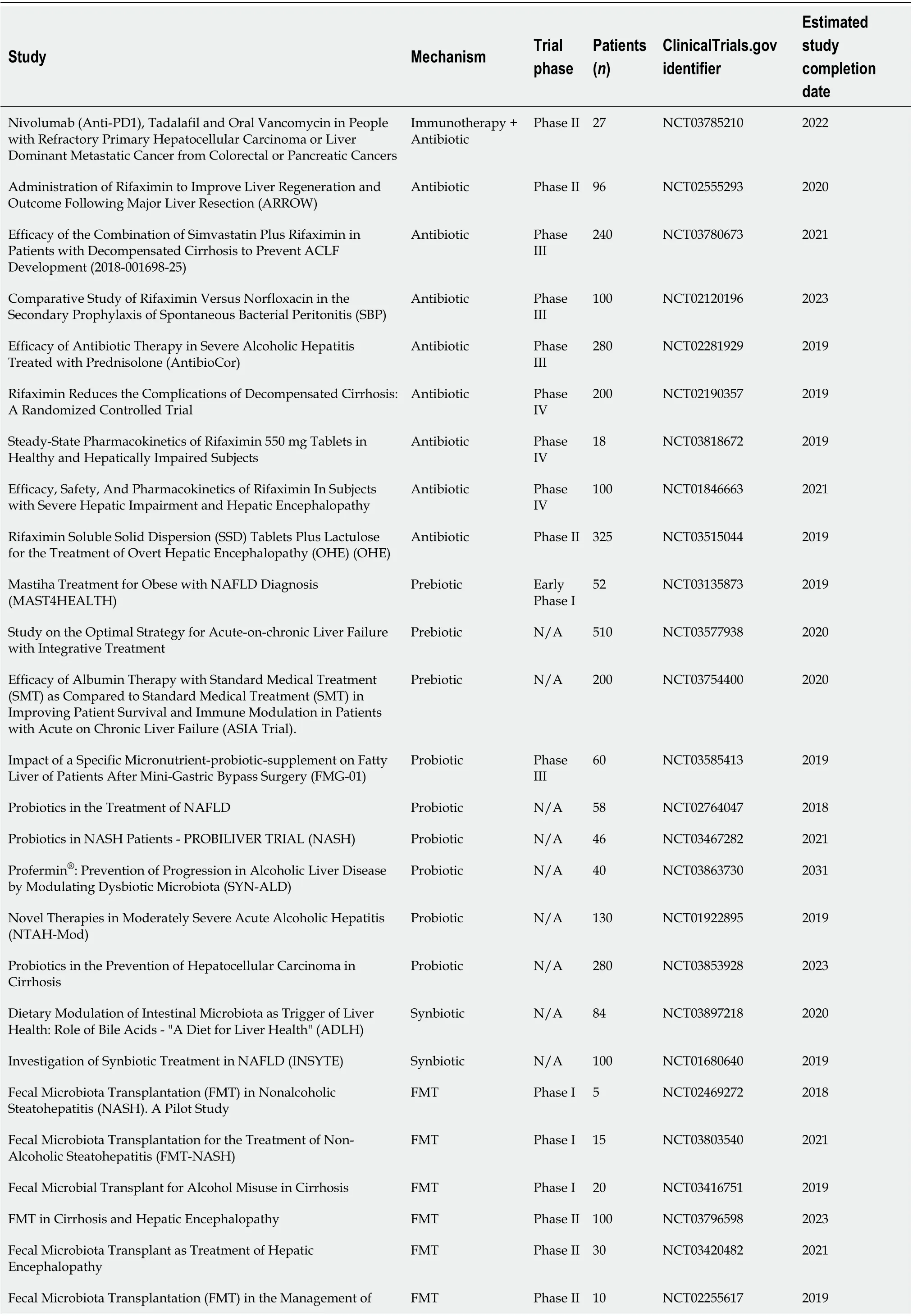
Table 2 Gut microbiota-based clinical trials

Hepatic Encephalopathy (HE): A Pilot Study Fecal Transplant for Hepatic Encephalopathy FMT Phase II 30 NCT03439982 2021 Trial of Faecal Microbiota Transplantation in Cirrhosis (PROFIT)FMT Phase III 32 NCT02862249 2019 To Assess the Role of Fecal Microbiota Transplant in Acute Liver Failure FMT N/A 40 NCT03363022 2018
杂志排行

World Journal of Gastroenterology的其它文章
- Research advances of vasoactive intestinal peptide in the pathogenesis of ulcerative colitis by regulating interleukin-10 expression in regulatory B cells
- Pretreatment with intestinal trefoil factor alleviates stress-induced gastric mucosal damage via Akt signaling
- Gegen Qinlian decoction enhances immunity and protects intestinal barrier function in colorectal cancer patients via gut microbiota
- Primary intestinal lymphangiectasia in an adult patient: A case report and review of literature
- Impact of colorectal cancer screening participation in remote northern Canada: A retrospective cohort study
- High prevalence of hepatic steatosis and vascular thrombosis in COVID-19: A systematic review and metaanalysis of autopsy data