Research advances of vasoactive intestinal peptide in the pathogenesis of ulcerative colitis by regulating interleukin-10 expression in regulatory B cells
2021-01-15XiongSunYaoHuangYaLiZhangDanQiaoYanChengDai
Xiong Sun, Yao Huang, Ya-Li Zhang, Dan Qiao, Yan-Cheng Dai
Abstract Ulcerative colitis (UC) is a chronic relapsed intestinal disease with an increasing incidence around the world. The pathophysiology of UC remains unclear. However, the role of the interaction between the enteric nervous system and the immune system in the pathogenesis of UC has been the focus of attention and has become a research hotspot. Vasoactive intestinal peptide (VIP) is a kind of endogenous neuropeptide with regulatory activity on intestinal immunity. It has been shown to regulate immune disorders in animal and human experiments and has become an effective anti-inflammatory and immune modulator that affects the innate immune system and adaptive immune system. Regulatory B cells (Bregs) are a new group of B cells that negatively regulate the immunity and have received extensive attention in immune circles. Bregs can regulate immune tolerance by producing interleukin (IL)-10, IL-35, and transforming growth factorβ, suppressing autoimmune diseases or excessive inflammatory responses. The secretion of IL-10 by Bregs induces the development of T helper (Th) 0 and Th2 cells. It also induces Th2 cytokines and inhibits Th1 cytokines, thereby inhibiting Th1 cells and the Th1/Th2 balance. With further clarity on the mechanism of the regulation of IL-10 expression by VIP in Bregs in colitis patients, we believe that Bregs can provide a novel strategy for the clinical treatment of UC. Thus, we aim to review the current literature on this evolving topic.
Key Words: Vasoactive intestinal peptide; Ulcerative colitis; Interleukin-10; Bregs; Pathogenesis
INTRODUCTION
Inflammatory bowel disease (IBD) is a global health problem. It consists of two main types, Crohn’s disease (CD) and ulcerative colitis (UC). UC is a chronic non-specific IBD that mainly involves the mucosa and submucosa of the rectum and colon. With a significant increase in the incidence of UC, it was identified as a modern refractory disease by the World Health Organization[1]. It is currently believed that UC is caused by an abnormal and continuous immune response to intestinal microorganisms and is facilitated by the genetic susceptibility of an individual[2].
The immune response of the human body includes innate and adaptive immunity. Innate immunity, which is the first defense of the body against pathogens, is nonspecific and does not provide lasting immunity; it is mediated by pattern recognition receptors, including toll-like receptors (TLRs) on the cell surface and nucleotidebinding oligomerization domain-like receptors in the cytoplasm[3]. Adaptive immunity is a highly specific and adaptive type of immunity. T helper (Th) cells are the key to an adaptive immune response. Most studies suggest that the pathological characteristics of UC are mainly an abnormal Th2 reaction, whereas an abnormal Th1 reaction is mainly found in CD[2,4]. Therefore, studying the underlying mechanism of intestinal mucosal immune response disorders can help us better understand the pathogenesis of IBD. Herein, the role of vasoactive intestinal peptide (VIP) and regulatory B cells (Bregs) in the pathogenesis of UC as well as the interaction between these two are reviewed, in the context of the immune pathogenesis of UC.
ROLE OF BREGS AND INTERLEUKIN-10 IN INTESTINAL IMMUNITY IN UC PATIENTS
A study found that dextran sulfate sodium (DSS) treated mice showed more severe colitis without B cells, and the filtration metastasis of B cells alleviated the disease. During colitis, the number of regulatory T cells (Tregs) of gut-associated lymphoid tissue (GALT) in B-cell deficient mice decreased significantly. After B cells were transferred into these mice, the number of GALT Tregs was restored, indicating that B cells contribute to Treg homeostasis. The study also found that B cells can induce the proliferation of Tregs and then promote the differentiation of B cells into IgAproducing plasma cells. These results suggest that B cells and Tregs interact and cooperate to prevent excessive immune response, which can lead to colitis[5]. Strong stimulation of B cells byEscherichia colileads to significant expression of inhibitory molecules on the surface of B cells and increases the production of anti-inflammatory cytokines, such as interleukin (IL)-10. Bregs induced by the bacterium can effectively inhibit the maturation and function of dendritic cells (DCs), inhibit the proliferation and polarization of Th1 and Th17 cells, and promote the differentiation of Th2 cells. In addition, Bregs promote the development of Tregs, which may establish immune homeostasis through feedback cooperation. The number of Bregs is directly related to the severity of inflammation. These findings may provide new methods for B-cell control in the treatment of autoimmune diseases[6].
IL-10 is an anti-inflammatory cytokine produced by monocytes, B cells, T cells, as well as some other cells. It is an important cytokine that induces the immune response of Th2 cells. IL-10 is expressed in different cells involved in innate and adaptive immunity, including dendritic cells, macrophages, T cells, natural killer cells, and B cells[7,8]. On the one hand, IL-10 protects the host from tissue damage caused by excessive inflammation, inhibits antigen presentation and production, and suppresses antigen expression and the production of pro-inflammatory chemokines and cytokines. IL-10 is the key gatekeeper in maintaining distal immune homeostasis. When specific pathogens enter the intestinal track, distal immune homeostasis may be imbalanced[9]. On the other hand, it enhances the survival, proliferation, differentiation, and homotypic transformation of human B cells[10]. The reduction of this anti-inflammatory cytokine is an important feature of UC[11].
IL-10 helps to maintain tissue integrity and promotes intestinal tissue homeostasis through its anti-inflammatory, anti-apoptotic, and tissue regeneration properties. The new immune cytokine, F8-IL-10 (Dekavil), a targeted antibody associated with IL-10, is currently being evaluated in phase 2 clinical trials in patients with rheumatoid arthritis, potentially paving the way for its use in IBD patients[12]. Mizoguchiet al[13]first used the concept of Bregs to define these B cell populations with negative regulatory functions[13]. Yanabaet al[14]defined the B cell subsets, CDld hiCD 5+, that secrete IL-10 in the spleen of mice; B10 cells do as well[14]. At present, the mechanism of Bregs inhibiting immune inflammation is realized by the following aspects: Bregs mainly exert an immune regulatory function through the secretion of IL-10 and transforming growth factor-β. IL-10 produced by Bregs affects the disease process by regulating T cell differentiation[15]. For example, in IBD, the Th1/Th2 balance can be regulated to suppress the harmful immune response[16]. Macrophages and DCs are also the main targets of IL-10 by playing an inhibitory and regulatory role, which can reduce the release of inflammatory factors from macrophages or monocytes and reduce the inflammatory response[17].
Bregs play a regulatory role through intercellular contacts. Studies have shown that Bregs promote the proliferation and activity of Tregs through direct major histocompatibility complex and B7 pathways and inhibit the proliferation of effector T cells through Fas/FasL-mediated apoptosis[18,19]. Studies on the interactions between Bregs and other regulatory immune cells have shown that co-culture of Bregs and CD4+T cells can quantitatively reduce the proliferative capacity of CD4+T cells[20]. B10 cells induced by mannose-capped lipoarabinomannan (ManLAM) were transferred into IL-10-/-mice, and B10 cells induced by ManLAM can reduce the severity of colitis in mice. B10 cells down-regulated Th1 polarization of spleen and mesenteric lymph nodes in DSS treated mice. These results suggest that the production of IL-10 by B cells treated with ManLAM helps to maintain the balance between CD4+T cell subsets and protects mice against DSS-induced IBD[21].
Bregs generate antibodies, neutralize harmful soluble factors, inhibit the activity of DCs/macrophages through the immunoglobulin G/FcTRII interaction, enhance the clearance of apoptotic cells, and reduce the potential autoantigen activating autoreactive T cells. In a recentin vitrostudy, the secretion of IL-10 and IL-12 p40 was significantly decreased and IL-12 p40 was increased in phosphoinositide 3-kinase (PI3K) kinase (d910a/d910a) mice or wild-type B cells treated with PI3K specific inhibitors. Macrophages or CD4+T cells activated by co-cultured microorganisms did not inhibit inflammatory cytokines.In vivo, co-transferred wild-type B cells improved T-cell-mediated colitis, while PI3K kinase (d910a/d910a) B cells had no protective effect on mucosal inflammation. These results suggest that the PI3K pathway plays an important role in the induction and regulation of IL-10 production in B cells in intestinal homeostasis and inflammation[22].
In humans, B-cell depletion using anti-CD20 (rituximab) for various diseases has been reported either to aggravate colitis or lead to spontaneous colitis[23,24]. Feces were transplanted into sterile (GF) IL10+/EGFP reporting mice and IL10-/-mice. It was proved that the microbiota in specific pathogen-free mice mainly stimulated IL-10 to produce colon-specific B cells and T-regulatory-1 cells. IL-10, in turn, down-regulated the microbial activated inflammatory cytokines. These studies suggest that resident intestinal bacteria activate IL-10 producing B cells through the TLR2, MyD88, and PI3K pathways. These B cells reduce the activation of colon T cells and maintain mucosal homeostasis under the action of intestinal microbiota[25]. The B cell subsets that produce IL-10 have different phenotypes and origins. Such B cell subsets appear in chronic inflammatory processes and inhibit the progression of intestinal inflammation by down-regulating the inflammatory cascade. In response to wounds, epithelial cells migrate and proliferate to cover the mucosal surface and repair barrier defects. This process is coordinated by immune cells and epithelial cells; however, the mechanism is not fully understood, and after mucosal injury, macrophage-derived IL-10 induces activation of epithelial cAMP response element binding protein (CREB) and then synthesis and secretion of Wnt1-inducible signaling protein 1 (WISP-1). WISP-1 induces epithelial cell proliferation and wound healing by activating the epithelial proliferation pathway. These findings suggest that macrophages are involved in regulation of the IL-10/CREB/WISP-1 signal axis and have broad significance in the relationship between innate immune activation and mucosal injury repair[26]. IL-10 is a key regulator of mucosal homeostasis. Some studies have shown that residents ofE. coli-induced chronic colitis in mice have a rapid but temporary activation effect on the immune system in normal hosts, and the induced protection of IL-10 B cells and CD4+cells subsequently suppresses this reaction and modulates homeostasis in the mucosa[27].
In summary, IL-10 is the key factor for the negative regulatory function of Bregs. However, further studies are required to clarify the function of IL-10 secreted from Bregs. Whether it is expressed by specific transcription factors and whether immune tolerance to autoantigens is temporary or permanent needs to be determined.
ROLE OF VIP IN INTESTINAL IMMUNITY IN UC PATIENTS
VIP is a 28 amino acid neuropeptide that was originally isolated from the small intestine of pigs in 1970 by Sami Said and Victor Mutt. It belongs to the neuropeptide family that includes the pituitary adenylate cyclase activated polypeptide (PACAP). In the gastrointestinal tract, VIP occurs in endocrine cells and neurons of the enteric nervous system[28]. In established CD models, such as the colitis model induced by enteral injection of trinitrobenzene sulfonic acid (TNBS), VIP reduces the clinical and histopathological severity of the disease and reduces weight loss, diarrhea, and macroscopic and microscopic intestinal inflammation[29]. VIP treatment reduced the levels of various chemokines and pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α), IL-6, IL-12, and IL-10[30]. In food allergy patients with enteritis, studies have found that VIP can increase the expression of TSP1, stabilize the expression of IL-10 in food allergy Bregs, and restore immune regulation function, thereby reducing the food allergy response. The results show that VIP has therapeutic potential for treating food allergy and other diseases related to Bregs immune regulation dysfunction[31].
However, the therapeutic effect of VIP in the management of UC has not been confirmed. Some studies have shown that the number of VIP positive nerve fibers in UC patients either decreases or remains unchanged[32-34]. Other studies have revealed that the level of VIP in the colon of UC patients increases with increased mRNA expression and VIP levels in neurons[34,35]. Similarly, plasma VIP level was also found to be elevated in UC patients[36]. Studies on the expression of VIP in UC patients are limited and contradictory, which might be due to the diversity of tissue sampling methods and patient populations. Jayawardenaet al[37]reported that the VPAC1 receptor might mediate the pro-inflammatory effect of VIP, and VPAC2 might mediate the anti-inflammatory effect of DSS-induced colitis[37]. In addition, recent studies have shown that VIP and its receptor, VPAC1, cannot be detected in the tissues of IBD patients with severe mucosal barrier disorders[38]. These results suggest that the VIP response disorder may be one of the causes of IBD.
VIP plays a key role in protecting the colon epithelium from pathogenic bacteria. Impaired crypt cell dynamics in VIP knockout mice, including reduced proliferation and migration of intestinal epithelial cells (IEC) and their increased apoptosis, resulted in a percolating intestinal barrier susceptible to DSS- and TNBS-induced colitis[13]. In addition, impaired proliferation and increased apoptosis of IEC may be one of the reasons for impaired epithelial regeneration in VIP knockout mice. Similarly, in IBD patients, the inflammatory changes in VIP+neurons and their receptors may lead to occurrence of the disease through the loss of VIP-mediated epithelial homeostasis regulation[32,38,39].
VIP plays a critical role in the development and maintenance of the integrity of colonic epithelium and the mucus barrier, possibly by activating caudal type homeobox transcription factor 2. VIP regulates the proliferation, migration, maturation, and secretion of bioactive cup-cell peptides in Cogu crypt cells to promote tissue repair and homeostasis, thereby controlling the susceptibility to colitis.
VIP REGULATES THE EXPRESSION OF IL-10 IN BREGS
VIP is a peptide secreted by nerve and immune cells. As a cytokine/chemokine, VIP performs a wide range of immune functions. It affects the innate and adaptive immunity through interactions with specific receptors, VPAC1, VPAC2, chemoattractant receptor-homologous molecule 2, and PAC1[40]. Both T and B lymphocytes express the VIP-related receptors[41], suggesting that B lymphocytes may regulate their immune response through VIP partly[42]. The levels of VIP in neurons and non-neurons were lower in UC patients. In UC, substance P and VIP decreased simultaneously. Further study is needed to determine the role of the neuropeptide in UC[43].
In innate immunity, VIP inhibits the production of inflammatory cytokines (interferon-γ, TNF-α, IL-6, and IL-17) and chemokines by immune cells (monocyte chemotactic protein-1, nuclear factor kappa-B), reduces the expression of costimulatory molecules (B7-1, B7-2 and CD40) on antigen-presenting cells, and reduces the stimulation of antigen-specific CD4+T cells. In addition, in adaptive immunity, VIP promotes the Th2 immune response and reduces the inflammatory Th1 response[44]. Although the exact mechanism remains to be clarified, VIP seems to regulate the Th1/Th2 balance in several ways as discussed below. First, VIP inhibits the production of the Th1-related cytokine, IL-12[45]. Second, VIP was reported to induce the expression of CD86 in resting mouse DCs, which played an important role in the development of Th2 cells[46]. Third, VIP inhibited CD95 (FasL) and granulase Bmediated apoptosis of mouse T2 cells but not of Th1 effector cells[47]. Lastly, VIP induced Th2 major transcription factors, c-MAF, GATA-3, and JUNB, during the differentiation of CD4+T cells in mice to inhibit T-bet, which is necessary for the differentiation of Th1 cells[48]. Therefore, VIP regulates the Th1/Th2 balance by directly acting on T cell differentiation and indirectly regulating the antigen presenting cell function.
Previous studies have shown that exogenous VIP can alleviate the histopathological symptoms of TNBS-induced colitis[29]. Studies have shown that rVIPa (VIP analogue) can improve TNBS-induced colitis in rats and effectively protect intestinal mucosal barrier function, and rVIPa can be used as a new alternative therapy for the treatment of intestinal inflammatory diseases[49]. VIP reinstated immune tolerance by downregulating the inflammatory response and inducing Tregs[50]. VIP and PACAP inhibit IL-10 through direct transcriptional events. Unlike IL-2, IL-4 and IL-10 act as proinflammatory or anti-inflammatory cytokines, depending on the type of immune response they are involved in[51]. Specific VPAC1 receptors mediate the stimulation of VIP/PACAP, and cAMP is the main second messenger involved. VIP/PACAP increased lipopolysaccharide (LPS) to stimulate IL-10 mRNA in cells, and the effects of transcription and protein synthesis inhibitors suggested thede novoproduction of IL-10 protein. It may work together with pro-inflammatory cytokines, such as IL-6 and TNF-α,in vivoto reduce the intensity of the immune response. It has been proved that lentivirus VIP can down-regulate the expression of pro-inflammatory TNF mRNA and protein and up-regulate the expression of anti-inflammatory IL-10 mRNA and protein. In addition, VIP reduced the expression of TNF in mouse macrophages stimulated by LPS through the protein kinase C and protein kinase A pathways.
CD5+B cells are regulatory immune cells. Some studies have found that the expression level of TLR9 in colonlamina propria(LP) CD5+B cells is higher than that in CD5- B cells. Compared with LP CD5- B cells, colon LP CD5+B cells stimulated by TLR ligand produced more IL-10. Acute intestinal inflammation can temporarily reduce the frequency of colon LP CD5+B cells, while chronic inflammation can lead to the continuous decrease of colon LP CD5+B cells and lead to the state of mainly CD5- B cells. This study concluded that the continuous changes in mucosal B cells caused by chronic intestinal inflammation may be involved in the pathogenesis of IBD[52]. However, IL-10 secreted by Bregs may also be regulated by VIP and may affect the immune balance in intestinal inflammation, but this needs to be confirmed in future studies[53]. Recently, we found in human peripheral blood samples and in animal experiments that insufficient VIP levels in the UC intestinal microenvironment accelerated the degradation of IL-10 mRNA, leading to dysfunction of Bregs[54]. VIP plays an important role in stabilizing the expression of IL-10 mRNA in Bregs. The administration of VIP can effectively inhibit experimental colitis in mice, suggesting the transformation potential of VIP in the treatment of UC.
In summary, VIP is a neuropeptide present in the lymphoid microenvironment and is a potent anti-inflammatory drug that inhibits the function of activated macrophages and Th cells. VIP may regulate the expression of IL-10 through different pathways, and IL-10 produced by B10 cells has been shown to play an important role in the Th2 immune regulation of UC. Stimulation of the transcription of IL-10 by VIP/PACAP could have important therapeutic potential (Figure 1). To date, VIP (Avidil in clinical application) has been successfully used in pulmonary hypertension and sarcoidosis. VIP in its native form, using sterically stabilized micelles treatment significantly reduced the mRNA levels of pro-inflammatory cytokines and showed significant histological recovery. Therefore, these results indicate that as a nanodrug, the antiinflammatory and anti-diarrhea effects of VIP can be achieved in a single dose. Therefore, the study suggested the development of VIP SSM as a potential therapeutic tool for UC[55].
CONCLUSION
Usually, peripheral blood samples are collected from UC patients. Bregs are isolated from these samples and their immune regulatory function (serum IL-10 and VIP levels) is analyzed. Although an increasing number of studies have focused on Bregs in recent years, many problems concerning Bregs still need to be resolved. It remains unclear which antigen can promote the development of Bregs. Moreover, although TLRs, CD40, and CD80/CD86 are known to be involved in the activation of Bregs, whether other important co-stimulatory molecules or cytokines are involved in this process is unclear. In addition, IL-10 is a key factor in the negative regulatory function of Bregs, and further studies are needed to determine how IL-10 secreted by Bregs plays its role, whether it is expressed by specific transcription factors and whether immune tolerance to autoantigens is temporary or permanent. With further clarity on the mechanism of the regulation of IL-10 expression by VIP-Breg axis of colitis patients, we believe that VIP would provide a novel strategy for the clinical treatment of UC.
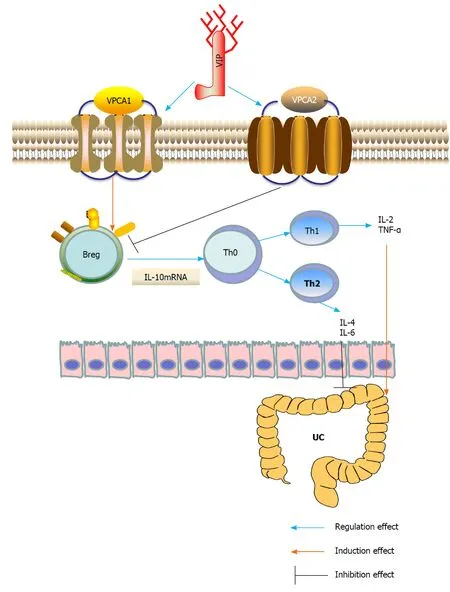
Figure 1 Underlying mechanisms of vasoactive intestinal peptide interference of ulcerative colitis by regulating the expression of interleukin-10 in regulatory B cells. Breg: Regulatory B cell; IL: Interleukin; Th: T helper; TNF-α: Tumor necrosis factor-α; UC: Ulcerative colitis; VIP: Vasoactive intestinal peptide.
杂志排行

World Journal of Gastroenterology的其它文章
- Gut microbiota mediated molecular events and therapy in liver diseases
- Pretreatment with intestinal trefoil factor alleviates stress-induced gastric mucosal damage via Akt signaling
- Gegen Qinlian decoction enhances immunity and protects intestinal barrier function in colorectal cancer patients via gut microbiota
- Primary intestinal lymphangiectasia in an adult patient: A case report and review of literature
- Impact of colorectal cancer screening participation in remote northern Canada: A retrospective cohort study
- High prevalence of hepatic steatosis and vascular thrombosis in COVID-19: A systematic review and metaanalysis of autopsy data