HPLC法同时测定复方磺胺间甲氧嘧啶钠粉中磺胺间甲氧嘧啶钠和甲氧苄啶的含量
2021-01-04梁劲康张桂君黎健业吴志玲黎乃添方炳虎
梁劲康,张桂君,黎健业,吴志玲,黎乃添,方炳虎
(广东温氏大华农生物科技有限公司,广东云浮 527400)
复方磺胺间甲氧嘧啶钠粉是由磺胺间甲氧嘧啶钠、甲氧苄啶和葡萄糖等辅料混合配制成的粉剂,主要用于革兰氏阳性和阴性敏感菌引起的感染,如呼吸道、消化道、泌尿道感染及球虫病等。根据《兽药质量标准》(2017年版)中复方磺胺间甲氧嘧啶钠粉的质量标准,应分别采用永停滴定法和紫外可见分光光度法测定磺胺间甲氧嘧啶钠和甲氧苄啶的含量[1]。但与高效液相色谱法相比,上述两种检测方法操作较为繁琐,而且测定结果误差较大。经查阅文献,已有研究学者报道了利用高效液相色谱法测定复方磺胺间甲氧嘧啶钠溶液和注射液中磺胺间甲氧嘧啶钠和甲氧苄啶的含量[2-4]。本文在上述文献方法的基础上,进一步优化其色谱条件,建立了可同时测定复方磺胺间甲氧嘧啶钠粉中磺胺间甲氧嘧啶钠和甲氧苄啶的高效液相色谱方法。该方法快速简便,结果准确可靠,可为复方磺胺间甲氧嘧啶钠粉的质量标准评价方法提供技术支持。
1 材料与方法
1.1 仪器 Waters e2695高效液相色谱系统配置2489 UV/Vis检测器(美国Waters公司);CPA225D型电子天平(德国Sartorius公司);Milli-Q超纯水仪(美国Millipore公司);SHZ-D(Ⅲ)循环式真空泵(巩义市予华仪器有限责任公司);KQ-100E型超声波清洗器(昆山市超声仪器有限公司)。
1.2 试药 磺胺间甲氧嘧啶对照品(含量:99.5%,批号:C0031610,中国兽医药品监察所);甲氧苄啶对照品(含量:99.8%,批号:H0161704,中国兽医药品监察所);复方磺胺间甲氧嘧啶钠粉(规格为100 g:磺胺间甲氧嘧啶钠10 g +甲氧苄啶2 g,批号:20180701,广东温氏大华农生物科技有限公司新兴化药分公司生产技术部自制);乙腈(色谱纯,德国Merck公司);磷酸(分析纯,江苏强盛功能化学股份有限公司);超纯水(实验室自制,符合GB/T 6682-1992规定的一级水要求)。
1.3 方法
1.3.1 标准储备液的配制 精密称取12.66 mg磺胺间甲氧嘧啶对照品和12.57 mg甲氧苄啶对照品,置于25 mL容量瓶中,加入适量乙腈,超声使药物完全溶解后,补加乙腈至刻度,摇匀,制得磺胺间甲氧嘧啶和甲氧苄啶混合标准储备液(磺胺间甲氧嘧啶的浓度为503.87 μg/mL;甲氧苄啶的浓度为501.79 μg/mL)。
1.3.2 供试品溶液的配制 精密称取0.25 g复方磺胺间甲氧嘧啶钠粉样品于50 mL容量瓶中,加入适量流动相,超声使样品全部溶解后,补加流动相至刻度,摇匀;精密移取1 mL至10 mL容量瓶中,流动相稀释至刻度,摇匀后,制得供试品溶液。供试品溶液经0.22 μm微孔滤膜滤过,续滤液用于液相色谱分析。另取0.25 g不含药物的空白辅料样品,同法处理,制得空白辅料溶液。
1.3.3 色谱条件 色谱条件为:ECOSIL 120-5-C18(250 mm×4.6 mm,5 μm)色谱柱;以乙腈-0.1%磷酸溶液(20∶80,V/V)为流动相;流速为1.0 mL/min;检测波长为270 nm;柱温为30 ℃;进样量为20 μL。以磺胺间甲氧嘧啶和甲氧苄啶的色谱峰计算,理论塔板数应在3000以上。
1.3.4 专属性试验 取1.3.1项磺胺间甲氧嘧啶和甲氧苄啶混合标准储备液进行适量稀释后,按1.3.3项的色谱条件进样分析,记录色谱图。另分别取1.3.2项的供试品溶液以及空白辅料溶液,同法操作,记录色谱图。
1.3.5 灵敏度试验 分别精密移取一定体积1.3.1项的磺胺间甲氧嘧啶和甲氧苄啶混合标准储备液于10 mL的容量瓶中,流动相定容,进行逐级稀释。稀释液按1.3.3项的色谱条件进样分析,以信噪比(S/N)为3的检测浓度为检测限(LOD),以信噪比(S/N)为10的检测浓度为定量限(LOQ)。
1.3.6 磺胺间甲氧嘧啶和甲氧苄啶线性范围 分别精密移取一定体积的1.3.1项的磺胺间甲氧嘧啶和甲氧苄啶混合标准储备液,流动相稀释,配得磺胺间甲氧嘧啶的质量浓度分别为100.77、50.387、10.077、5.0387、1.0077、0.10077 μg/mL以及甲氧苄啶的的质量浓度分别为100.36、50.179、10.036、5.0179、1.0036、0.10036 μg/mL混合对照品稀释液。上述稀释液分别经0.22 μm微孔滤膜滤过后,取续滤液按1.3.3项下的色谱条件进样,记录峰面积。
1.3.7 精密度试验 分别精密移取一定体积的1.3.1项下的磺胺间甲氧嘧啶和甲氧苄啶混合标准储备液,用流动相稀释至磺胺间甲氧嘧啶的质量浓度分别为50.387、40.310和25.194 μg/mL以及甲氧苄啶的质量浓度分别为50.179、40.143、25.090 μg/mL的高、中、低3种混合对照品溶液。3种浓度的混合对照品溶液按1.3.3项下的色谱条件,连续重复进样5针,记录峰面积。
1.3.8 加标回收率试验 分别精密移取一定体积的1.3.1项下的磺胺间甲氧嘧啶和甲氧苄啶混合标准储备液以及1 mL 1.3.2项下的空白辅料溶液于10 mL容量瓶中,流动相稀释定容,摇匀,配得磺胺间甲氧嘧啶的质量浓度分别为50.387、40.310、25.194 μg/mL以及甲氧苄啶的质量浓度分别为50.179、40.143、25.090 μg/mL的高、中、低3种混合对照品溶液。每一浓度平行配制3份。上述溶液经0.22 μm微孔滤膜滤过后,取续滤液按1.3.3项下的色谱条件进样,记录峰面积,依据标准曲线计算加标回收率。
1.3.9 稳定性试验 取1.3.7项下的高、中、低3种磺胺间甲氧嘧啶和甲氧苄啶混合对照品溶液,分别于0、2、4、6、8、12、24 h按1.3.3项下的色谱条件进样分析,记录峰面积。
1.3.10 样品含量测定 平行称取5份复方磺胺间甲氧嘧啶钠粉供试品,按1.3.2项下的供试品溶液的配制方法同法操作,所得供试品溶液按1.3.3项下的色谱条件进样分析,记录峰面积,依据标准曲线计算磺胺间甲氧嘧啶钠和甲氧苄啶的含量。另外,平行称取5份供试品,按照《兽药质量标准》(2017年版)中复方磺胺间甲氧嘧啶钠粉项下的测定方法[1],分别采用永停滴定法和紫外可见分光光度法测定磺胺间甲氧嘧啶钠和甲氧苄啶的含量。
2 结果与分析
2.1 色谱条件的优化及确定 参考文献的色谱方法[2-5],在前期实验中对磺胺间甲氧嘧啶和甲氧苄啶的液相色谱条件进行了优化。当选用0.2%冰醋酸溶液以不同比例与乙腈组成的流动相进行洗脱时,磺胺间甲氧嘧啶和甲氧苄啶的色谱峰均出现了不同程度的拖尾现象;而当流动相组成为乙腈-0.1%磷酸溶液(20∶80,V/V)时,磺胺间甲氧嘧啶和甲氧苄啶的色谱峰峰型较好,因此,选择了乙腈-0.1%磷酸溶液(20∶80,V/V)作为磺胺间甲氧嘧啶和甲氧苄啶液相检测的流动相。此外,当选用长度为150 mm的色谱柱进行洗脱时,甲氧苄啶和磺胺间甲氧嘧啶的保留时间分别是1.5 min和4.5 min左右。为了提高两种药物色谱峰的分离度,本文最终选择了长度为250 mm的C18柱进行洗脱。
经优化后,确定了磺胺间甲氧嘧啶和甲氧苄啶的高效液相色谱检测条件为采用ECOSIL 120-5-C18(250 mm×4.6 mm,5.0 μm)色谱柱,在柱温为30 ℃条件下以乙腈-0.1%磷酸溶液(20∶80,V/V)为流动相按1.0 mL/min流速进行等度洗脱。在上述色谱条件下,磺胺间甲氧嘧啶和甲氧苄啶能得到更充分的分离,峰型更理想。
2.2 专属性试验 磺胺间甲氧嘧啶和甲氧苄啶的专属性实验结果可见图1。由图1中可以看出,空白辅料在该色谱条件下并不会干扰磺胺间甲氧嘧啶和甲氧苄啶的色谱峰。甲氧苄啶的出峰时间为5.6 min,而磺胺间甲氧嘧啶的出峰时间为13.6 min。两种药物的峰型较好,且分离度良好。
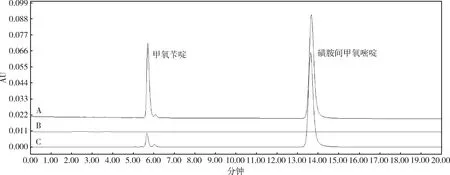
(A:磺胺间甲氧嘧啶和甲氧苄啶标准混合溶液;B:空白辅料溶液,C:供试品溶液。A for the mixed standard solution of the sulfamonomethoxine and trimethoprim;B for the blank excipient solution;C for the sample solution)图1 空白辅料、混合标准品溶液及供试品溶液的高效液相色谱图Fig 1 The HPLC spectrum of sulfamonomethoxine and trimethoprim blank, mixture standard and sample solution
2.3 灵敏度试验 实验结果表明,磺胺间甲氧嘧啶的LOD可达20.155 μg/L,LOQ则为50.387 μg/L;而甲氧苄啶LOD可达10.036 μg/L,LOQ则为100.36 μg/L。
2.4 磺胺间甲氧嘧啶和甲氧苄啶线性范围 利用1.3.3项下的高效液相色谱方法测得磺胺间甲氧嘧啶和甲氧苄啶的峰面积后,分别以峰面积A为纵坐标,质量浓度C为横坐标,进行线性回归。结果表明磺胺间甲氧嘧啶在0.10077~100.77 μg/mL范围内线性关系良好,回归方程为A1= 69756C1- 16072(R2=0.9998);甲氧苄啶在0.10036~100.36 μg/mL范围内线性关系良好,回归方程为A2= 24183C2- 2830.4(R2=0.9998)。
2.5 精密度试验 实验结果表明,磺胺间甲氧嘧啶在质量浓度为50.387、40.310和25.194 μg/mL时连续进样5针后,峰面积的RSD分别为0.19%、0.65%和0.20%。甲氧苄啶在质量浓度为50.179、40.143、25.090 μg/mL时连续进样5针后,峰面积的RSD分别为0.28%、0.87%和0.21%。上述结果均符合方法学要求,表明本方法精密度良好。
2.6 加标回收率试验 加标回收率试验的实验结果可见表1和表2。结果表明,磺胺间甲氧嘧啶在50.387、40.310、25.194 μg/mL等3种水平浓度下的平均加标回收率为100.03%、99.46%和98.68%,RSD分别为0.69%、0.46%和0.41%。甲氧苄啶在50.179、40.143、25.090 μg/mL等3种水平浓度下的平均加标回收率为99.95%、99.40%和98.86%,RSD分别为0.12%、0.85%和0.32%。上述结果均能满足方法学的要求,说明该方法准确度良好。
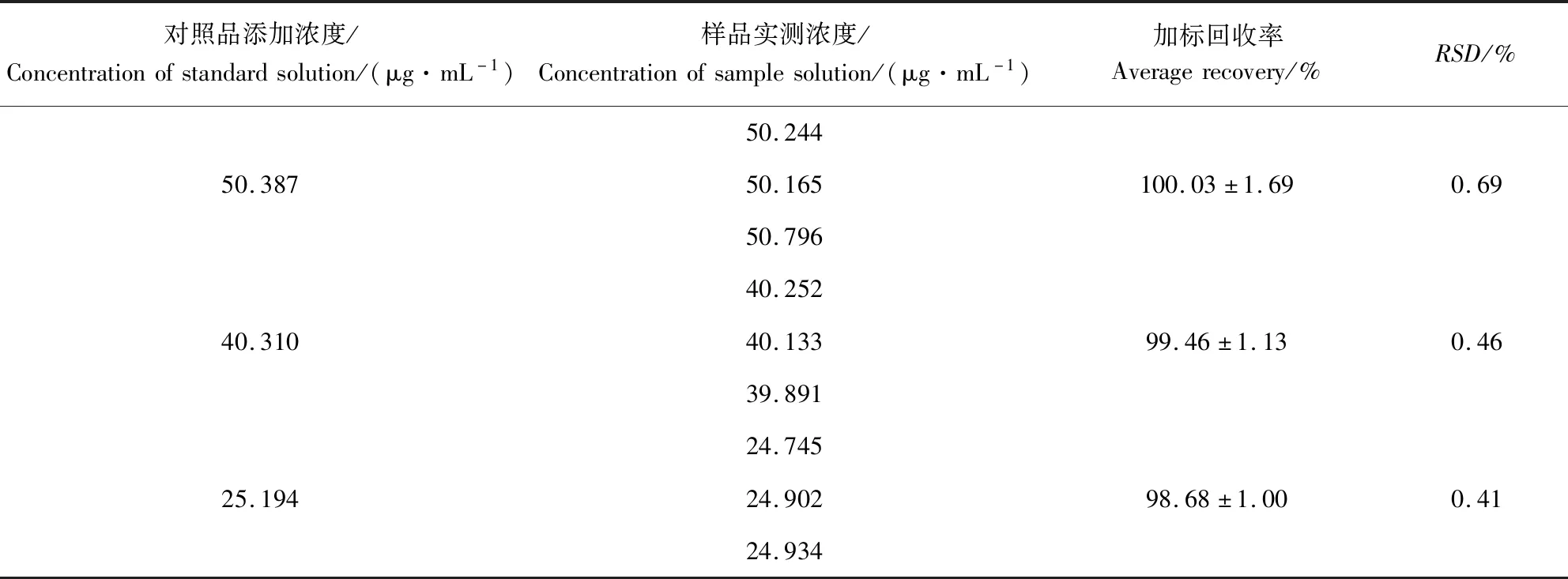
表1 磺胺间甲氧嘧啶加标回收率结果Tab 1 The average recovery results of sulfamonomethoxine
2.7 稳定性试验 稳定性试验的实验结果表明,磺胺间甲氧嘧啶在浓度为25.194、40.310和50.387 μg/mL时24 h内峰面积RSD分别为0.19%、0.11%和1.16%;甲氧苄啶在浓度为25.090、40.143、50.179 μg/mL时24 h内峰面积RSD分别为0.22%、0.39%和和1.08%,表明磺胺间甲氧嘧啶和甲氧苄啶在24 h内稳定性良好。
2.8 样品含量测定 供试品中磺胺间甲氧嘧啶钠和甲氧苄啶的测定结果可见表3和表4。5份供试品中磺胺间甲氧嘧啶钠和甲氧苄啶的含量均在标示量的90.0%~110.0%范围内,因此,5份供试品中磺胺间甲氧嘧啶钠和甲氧苄啶的含量均符合要求。分别将表2和表3中的实验结果通过SPSS Statistics 19软件进行t检验。结果表明,利用高效液相色谱法和永停滴定法测定磺胺间甲氧嘧啶钠含量的结果并无显著性差异(P>0.05),而利用高效液相色谱法和紫外分光光度法测定甲氧苄啶含量的结果也并无显著性差异(P>0.05)。
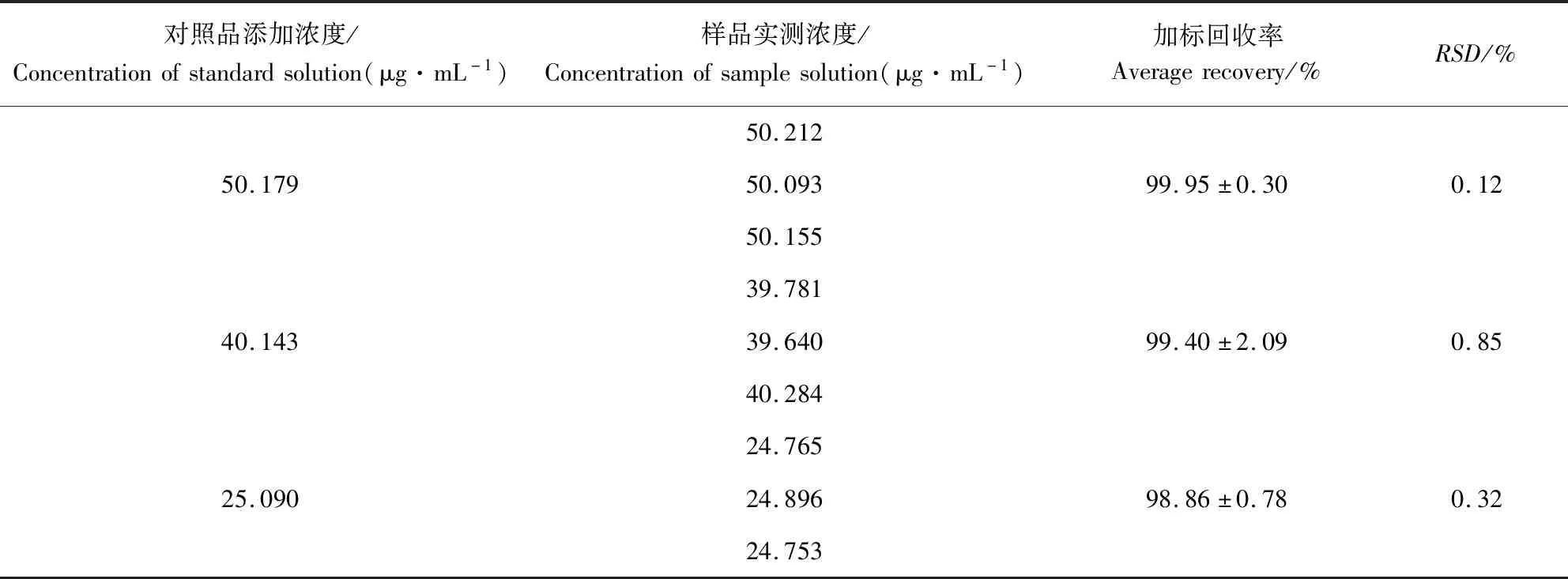
表2 甲氧苄啶加标回收率结果Tab 2 The average recovery results of trimethoprim
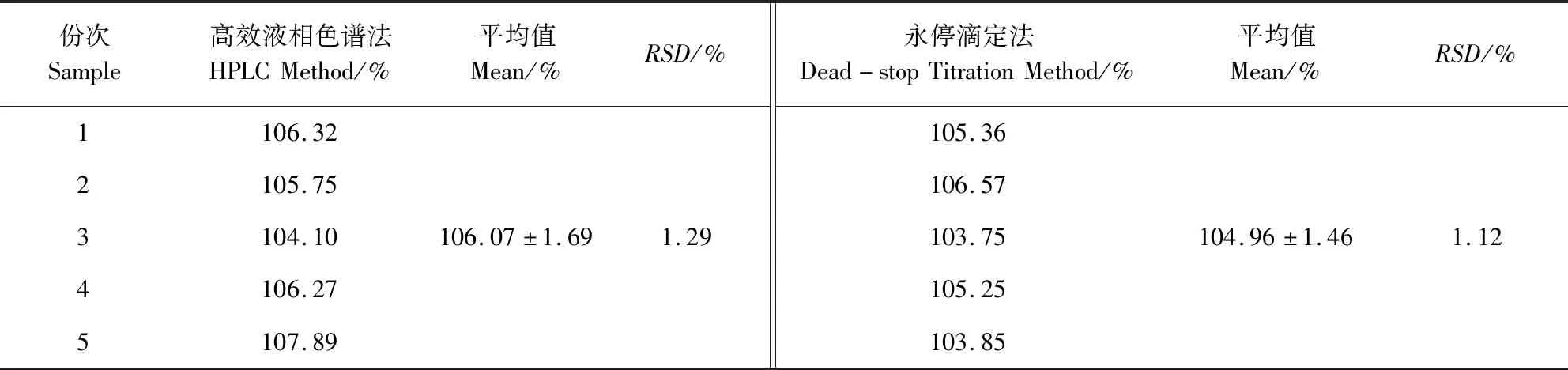
表3 磺胺间甲氧嘧啶钠的含量测定结果Tab 3 Results of the determined content of sulfamonomethoxine sodium
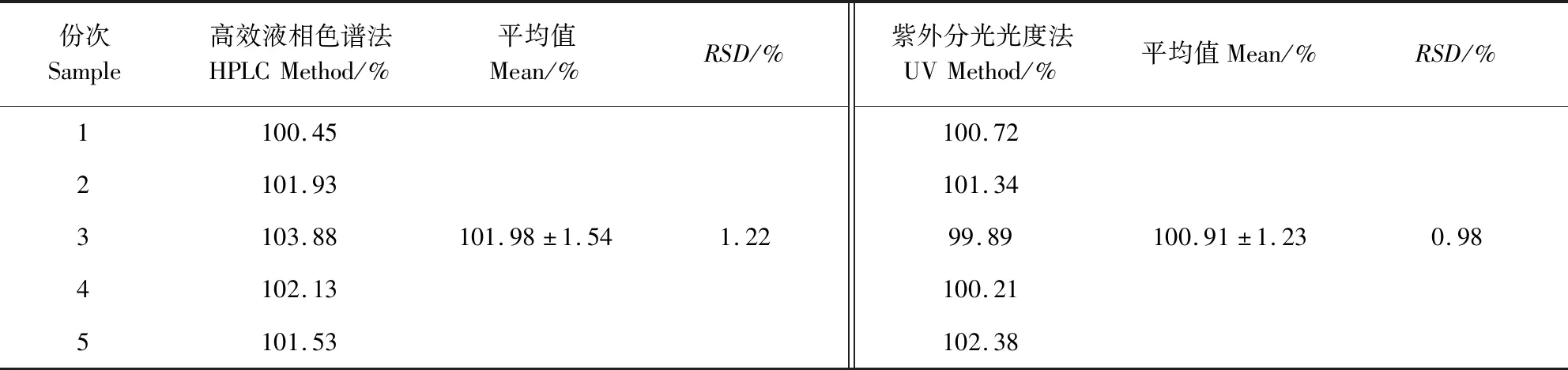
表4 甲氧苄啶的含量测定结果Tab 4 Results of the determined content of trimethoprim
3 讨论与结论
3.1 检测方法的比较 虽然国家标准中规定了分别采用永停滴定法和紫外可见分光光度法测定磺胺间甲氧嘧啶钠和甲氧苄啶的含量,但这两种测定方法操作繁琐复杂,极易引入测量误差。在采用永停滴定法测定磺胺间甲氧嘧啶钠的含量时,亚硝酸钠滴定液浓度及其消耗体积、移液管和滴定管校准、称量和基准物纯度等因素均是测量结果不确定度的主要来源,而且这些因素在实际操作中也很难以规避[6]。而高效液相色谱法测定磺胺间甲氧嘧啶钠含量引入不确定度影响因素主要是称量、对照品含量、溶液定容和重复进样等[7]。与永停滴定法相对扩展不确定度(U=0.8)相比,高效液相色谱法的相对扩展不确定度更小(U=0.5),也就说明了高效液相色谱法测定磺胺间甲氧嘧啶钠含量的准确度更高[6-7]。同样,采用紫外分光光度法测定甲氧苄啶的含量时,需先后使用三氯甲烷和稀醋酸进行提取后才能测定药物的含量。在提取过程中,极易造成药物的损失。而高效液相色谱法克服了原方法操作繁琐复杂的缺点,避免了操作过程人为因素对结果造成的影响[1,8]。因此,高效液相色谱法是较理想的检测磺胺间甲氧嘧啶钠和甲氧苄啶含量的方法之一。
3.2 检测波长的选择 为了进一步优化其高效液相色谱条件,本文也对磺胺间甲氧嘧啶钠和甲氧苄啶的检测波长和流动相组成等因素进行了筛选。杨学武等选择了230 nm作为磺胺间甲氧嘧啶钠和甲氧苄啶的液相检测波长[4]。但是,230 nm波长处接近于紫外末端吸收,容易引起基线漂移等问题。分别采用磺胺间甲氧嘧啶和甲氧苄啶对照品溶液在200~600 nm波长范围内进行扫描后发现,磺胺间甲氧嘧啶的紫外最大吸收波长为273 nm,甲氧苄啶紫外最大吸收波长为 271 nm,而二者在270 nm下均有较强吸收,因此,本文最终选择了检测波长为270 nm,与文献报道所选择检测波长一致[2-3]。
3.3 流动相的选择 张志美和郭时金等在磺胺间甲氧嘧啶钠和甲氧苄啶的液相分析检测中,采用了甲醇作为有机相进行洗脱[2-3]。但与甲醇相比,乙腈的极性更低,更有利于改善磺胺间甲氧嘧啶钠和甲氧苄啶分离度。此外,乙腈的紫外吸光度值比甲醇更低,能够避免溶剂的基质效应;而且等比例的乙腈与水混合后柱压也比甲醇更稳定,能够避免基线的波动,提高检测的准确性。因此,本文优先选择了乙腈作为流动相的有机相。由于磺胺间甲氧嘧啶钠和甲氧苄啶结构中均含有氨基基团,在流动相中加入适量的酸可避免峰型的拖尾现象。在前期实验中,本文也比较了冰醋酸、磷酸及其不同浓度对磺胺间甲氧嘧啶钠和甲氧苄啶的分离效果和对药物峰型的影响,其中以乙腈-0.1%磷酸溶液(20∶80,V/V)时,磺胺间甲氧嘧啶和甲氧苄啶的色谱峰峰型更好,分离度也更优。
3.4 色谱柱的选择 除了检测波长和流动相等因素外,色谱柱的长度对磺胺间甲氧嘧啶钠和甲氧苄啶的分离效果也具有较显著的影响。经筛选后,本文选择了长度为250 mm的C18柱进行洗脱,以提高两种药物色谱峰的分离度。
本文建立的高效液相色谱法能够使磺胺间甲氧嘧啶钠和甲氧苄啶充分分离,而且该方法简便有效,线性关系良好,精确度和灵敏度高,可作为复方磺胺间甲氧嘧啶钠粉中磺胺间甲氧嘧啶钠和甲氧苄啶同时定量的分析方法。
