可可毛色二孢全基因组分泌蛋白的预测及分析
2021-01-04邢启凯李铃仙曹阳张玮彭军波燕继晔李兴红
邢启凯,李铃仙,曹阳,张玮,彭军波,燕继晔,李兴红
可可毛色二孢全基因组分泌蛋白的预测及分析
邢启凯1,李铃仙1,曹阳2,张玮1,彭军波1,燕继晔1,李兴红1
(1北京市农林科学院植物保护环境保护研究所/北方果树病虫害绿色防控北京市重点实验室,北京 100097;2大连理工大学生物工程学院,辽宁大连 116024)
【】可可毛色二孢()是一种世界性分布的重要植物病原真菌,可引起严重的葡萄溃疡病(Botryosphaeria dieback),影响果木品质并造成巨大的经济损失。本研究预测并分析可可毛色二孢基因组范围内的分泌蛋白,并明确其基本特征,为该病菌分泌蛋白致病机理的研究打下基础。依据已公布的可可毛色二孢全基因组序列,利用信号肽预测软件SignalP v5.0、跨膜结构分析软件TMHMM v2.0、细胞器定位分析软件ProtComp v9.0、GPI锚定预测软件big-PI Fungal Predictor和亚细胞器定位分析软件TargetP v2.0生物信息学软件对该菌中的典型分泌蛋白进行筛选。对分泌蛋白N端信号肽的长度、氨基酸使用频率及其切割位点进行统计分析。依据蛋白序列的同源性,应用BLASTP程序对分泌组蛋白进行功能注释分析,预测其生物学功能。采用蔗糖酶缺陷的酵母分泌系统,对所选分泌蛋白的信号肽进行活性检测。利用qRT-PCR方法检测所选分泌蛋白基因在可可毛色二孢侵染葡萄中的表达情况。在可可毛色二孢全基因组编码蛋白中共筛选获得552个潜在的具有典型信号肽的分泌蛋白,占全基因组预测蛋白总数的4.3%,其编码蛋白长度集中于101—400 aa。信号肽统计分析表明,其信号肽长度以18—20 aa的序列最为集中,信号肽长度为20 aa的蛋白数量最多。信号肽中使用频率最高的氨基酸为丙氨酸;非极性、疏水的氨基酸使用频率最高,占氨基酸总数的60.2%。其信号肽的-3至-1位置上的氨基酸相对保守,切割位点属于A-X-A类型,可被Sp I型信号肽酶识别并切割。336个分泌蛋白具有功能注释,其功能较多集中于细胞壁降解有关的酶类以及致病相关蛋白,并且这些蛋白在分子量、等电点、脂肪族氨基酸指数等方面均存在差异。通过蔗糖酶缺陷的酵母分泌系统证实,挑选的9个分泌蛋白信号肽均具有分泌活性。qRT-PCR检测结果表明,所选分泌蛋白基因在该病菌侵染初期的表达发生变化。利用生物信息学分析技术从可可毛色二孢全基因组中共预测获得552个经典分泌蛋白。其信号肽氨基酸长度分布广泛,氨基酸组成中非极性、疏水的氨基酸使用频率最高。功能注释主要集中在细胞壁组分降解相关的酶类、致病侵染相关的坏死诱导相关蛋白以及几丁质结合蛋白等。
葡萄溃疡病;可可毛色二孢;生物信息学;分泌蛋白;信号肽;表达模式
0 引言
【研究意义】葡萄溃疡病(Botryosphaeria dieback)是葡萄()生产上最重要的枝干病害之一,在世界葡萄主产区造成巨大的经济损失[1]。通常认为,该类型病原真菌主要通过自然孔口或修剪伤口侵入,感病果木出现枝干溃疡、果梗干枯、果实干缩、掉粒以及树势减弱等症状,更严重的会引起根腐,造成整株果木枯死[2]。至今,在我国葡萄产区共分离获得6种葡萄座腔菌科(Botryosphaeriaceae)真菌可引起葡萄溃疡病,其中优势种群为可可毛色二孢()和葡萄座腔菌(),而致病力最强的为可可毛色二孢[3-5]。目前普遍使用化学药剂防治葡萄溃疡病,但至今没有获得低毒环保并能够高效防治该病害的有效药剂。病原真菌分泌蛋白(secreted protein)在病原真菌与宿主植物的互作中起着至关重要的作用,其直接影响病原菌的侵入、扩展、定殖以及病害发生[6-7]。可可毛色二孢全基因组范围内分泌蛋白的筛选鉴定,将对揭示其致病机理,进一步制定葡萄溃疡病的防治新策略具有重要意义。【前人研究进展】分泌蛋白是指在病原菌体内合成后,通过内质网/高尔基体等分泌途径转运到宿主细胞质膜外空间或细胞内发挥功能的蛋白分子[8-9]。经典的分泌蛋白具有以下特征[10]:(1)氨基酸N端含有信号肽序列;(2)无糖基磷脂酰肌醇(glycosylphosphatidylinositol,GPI)锚定位点;(3)没有跨膜结构域;(4)没有将蛋白输送至叶绿体、线粒体等胞内细胞器的定位信号。研究表明,在病原真菌的侵入、扩展和定殖等致病过程中,往往有大量的分泌蛋白参与其中,扮演着重要角色[11-12]。例如,在侵入宿主植物期间,病原真菌可以分泌大量的植物细胞壁降解酶、蛋白水解酶和代谢相关酶等酶类以及激发子蛋白,进而降解植物细胞壁以及植物细胞内复杂的碳氮化合物,一方面获得营养物质供病原菌生长,促使其在宿主中的侵入、定殖和扩散;另一方面阻碍或抑制宿主植物的免疫系统,从而完成其致病过程[9,13]。病原物来源的分泌蛋白与宿主植物受体蛋白的识别和信号传导研究是揭示病原物与宿主互作机制的关键点[14-15],因而研究者完成了多种病原菌全基因组水平的分泌蛋白预测研究[16-21],为其他病原菌分泌蛋白的预测及分析提供了参考。【本研究切入点】目前,关于葡萄溃疡病的研究主要集中在病原真菌的种类鉴定、种群结构、侵染过程以及毒素分泌等方面,关于其病原菌分泌蛋白的研究尚未报道。实验前期首先应用SOAPdenovo组装软件对可可毛色二孢菌株CSS-01s进行了测序组装分析[13],其全基因组大小为43.3 M,测序深度为90X,GC含量为54.77%[22]。通过组装拼接共获得60条contigs和29条scaffolds,并且通过基因软件预测并与各数据库比对共注释获得12 902个蛋白序列[22]。【拟解决的关键问题】在已公布的可可毛色二孢菌株CSS-01s全基因组信息的基础上[22],根据分泌蛋白典型特征,利用生物信息学在线程序进行预测分析,以期获得所有可编码经典分泌蛋白的相关基因,为揭示葡萄与可可毛色二孢等葡萄座腔菌科真菌互作的分子机制,并进一步持续有效地开展葡萄溃疡病的防控打下基础。
1 材料与方法
试验于2018—2019年在北京市农林科学院完成。
1.1 供试材料
供试可可毛色二孢菌株CSS-01s为课题组分离、单孢纯化、鉴定并保存。可可毛色二孢12 902个蛋白序列来源于PRJNA339237(accession number SRP107819)[22]。一年生葡萄‘夏黑’(Summer Black)绿枝条由北京市林业果树科学研究院提供。酵母信号序列诱捕载体pSUC2T7M13ORI(pSUC2)、对照载体和酵母菌株YTK12(suc2)由中国农业大学植物保护学院孙文献教授提供。
1.2 分泌蛋白筛选程序
应用SignalP v5.0[23](http://www.cbs.dtu.dk/services/ SignalP/)软件在线分析可可毛色二孢全基因组的蛋白序列是否含有N端信号肽以及该信号肽的切割位置;利用Protcomp v9.0[24](http://www.softberry.com/)在线分析软件进行亚细胞定位,获得其在亚细胞器的定位和分布;利用在线TMHMM v2.0[25](http://www.cbs. dtu.dk/services/TMHMM/)软件对候选序列的跨膜结构域进行分析;利用在线软件big-PI Fungal Predictor[26](http://mendel.imp.ac.at/gpi/fungi_server.html)实现蛋白质的GPI锚定修饰位点的预测;使用在线程序TargetP v2.0[27](http://www.cbs.dtu.dk/services/TargetP/)进一步分析靶标蛋白在亚细胞器中的定位和分布情况,以排除非胞外蛋白;利用LipoP v1.0[28](http:// www.cbs.dtu.dk/services/LipoP/)在线软件对分泌蛋白进行信号肽酶识别位点的预测;利用ExPAS[29](http://web.expasy.org/protparam/)在线软件预测分泌蛋白的理化性质。
1.3 分泌蛋白信号肽功能分析
1.3.1 载体构建 参照Jacobs等[30-31]方法,用蔗糖酶缺陷的酵母分泌系统对预测的分泌蛋白信号肽的功能进行分析验证。根据可可毛色二孢效应子序列信息,用生物信息学在线程序SignalP v5.0进行预测分析所选效应子的信号肽区域,信号肽通常是位于蛋白质N端、大小为40个氨基酸的多肽。依据该目的区段,利用Primer5.0在线程序设计载体构建所需的特异性引物,并在引物5′和3′端分别添加R I和I酶切位点,保证目的片段可单方向插入功能质粒。Trizol(Invitrogen)法提取可可毛色二孢的RNA,反转录得到cDNA。以此为模板进行PCR扩增,对表1所选的9个候选分泌蛋白信号肽片段进行扩增,反应程序:95℃预变性3 min;进入95℃变性30 s,60℃退火30 s,72℃延伸20 s,共32个循环;72℃延伸10 min。所用引物见表1,引物由上海生工生物工程有限公司合成。用R I和I酶切PCR片段,将回收产物连接到pSUC2相同的酶切位点间,随后对克隆进行PCR筛选鉴定,最后将重组质粒送北京擎科新业生物技术有限公司进行测序验证。
1.3.2 酵母感受态的制备与载体转化 依照Frozen- EZ Yeast Transformation II kitTM(Zymo Research,Orange,CA,USA)提供的方法制备蔗糖酶缺陷型酵母YTK12菌株的感受态,具体方法参照试剂盒说明书。将酵母YTK12菌株在YPDA平板上划线,30℃培养3—5 d;挑单斑至YPDA液体培养基摇培,直到OD600=0.7—0.8,收集菌液,用E1悬浮;再次离心倒掉上清,加入500 μL的E2,即为制备好的YTK18感受态,-80℃保存备用。
将构建好的载体、阳性对照载体pSUC2-Avr1b以及阴性对照载体pSUC2-Mg87分别转化到酵母YTK18感受态中,涂到CMD-W培养基(0.67%不含氨基酸的酵母氮源、0.075%色氨酸一缺培养基、0.1%葡萄糖、2%蔗糖和2%琼脂)上培养3—5 d。将筛选获得的转化子划线到YPRAA培养基(1%酵母提取物、2%蛋白胨、2 μg·L-1的抗霉素A、2%棉籽糖和2%琼脂)上培养3—5 d,通过观察酵母菌在YPRAA上的生长状况来判断所选分泌蛋白的信号肽是否具有分泌功能。

表1 分泌蛋白信号肽活性测定载体构建引物序列
1.4 可可毛色二孢侵染下分泌蛋白基因的表达分析
参考Yan等[3,22]接种葡萄座腔病菌的方法,用可可毛色二孢CSS-01s菌株接种一年生葡萄‘夏黑’绿枝条,于接种后0、6和12 h取接种点0.5—2 cm葡萄枝条表皮,液氮冻存后于-80℃冰箱保存备用。组织经液氮充分研磨后,用EASYspin Plus多糖多酚复杂植物RNA提取试剂盒(艾德莱生物)提取总RNA,经DNase I(TaKaRa)处理后,用SuperScript III反转录酶(Invitrogen)反转录得到第一链cDNA,-20℃保存,用于后续的实时荧光定量PCR分析(qRT-PCR)。依据全基因组预测的分泌蛋白序列设计特异性引物(表2),以(KAB2581229.1)作为内参基因,进行qRT-PCR分析。采用SYBR Green I荧光染料法,试验过程参照TaKaRa说明书。20 μL反应体系:2×TB GreenII(Tli RNaseH Plus)10 μL,正反引物(0.5 μmol·L-1)3.5 μL,cDNA 0.5 μL,ROX Reference Dye II 0.5 μL,补水至总体积为20 μL。qRT-PCR在ABI7500仪器上进行。反应程序:95℃ 5 min;进入95℃ 30 s,60℃ 30 s,共45个循环。每个反应得到相应的Ct值。每个样品进行3个平行反应,取均值后,用2-ΔΔCt方法计算基因的相对表达量。
2 结果
2.1 可可毛色二孢全基因组水平的分泌蛋白筛选鉴定
基于可可毛色二孢全基因组测序数据[22],对12 902条蛋白序列,通过SignalP v5.0[23]预测得到937个在氨基端含有典型信号肽序列的蛋白,占全基因组蛋白序列总数的7.26%。随后以ProtComp v9.0[24]在线程序对这937个含有信号肽的蛋白序列的细胞定位进行预测分析,结果如图1所示,共有685个蛋白可转运至细胞外,是胞外分泌蛋白类型,占比73%;另有252个蛋白未分泌到细胞外,其中最多的72个蛋白序列转运至细胞质膜(7.7%),其次是转运至线粒体(7.5%)、溶酶体(3%)和细胞质(2.9%);另外转运至过氧化酶体、细胞核、内质网和液泡的共有39个,占比4.2%。

表2 实时荧光定量PCR所用引物序列
为了排除含有跨膜结构域的蛋白序列,通过TMHMM v2.0在线软件[25]对氨基端含有典型信号肽的蛋白序列进行跨膜螺旋结构分析,结果如图2所示,该685个蛋白序列中,有576个蛋白序列不含跨膜结构域,占比84.1%;有98个蛋白序列只含有1个跨膜结构域,占比14.3%;其余11个蛋白序列含有2—3个跨膜结构域,占比1.6%。
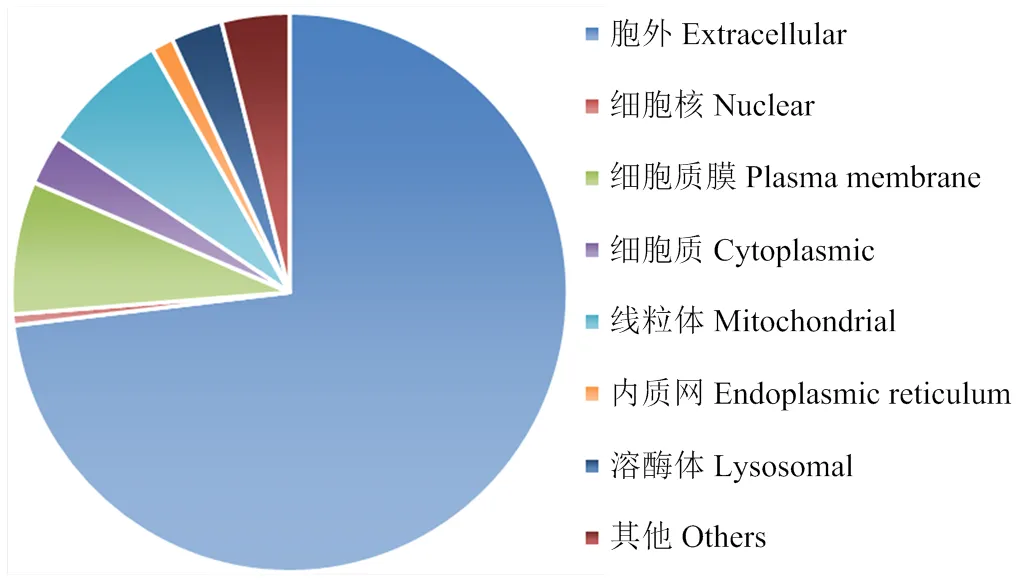
图1 可可毛色二孢中937个具有信号肽蛋白的亚细胞定位
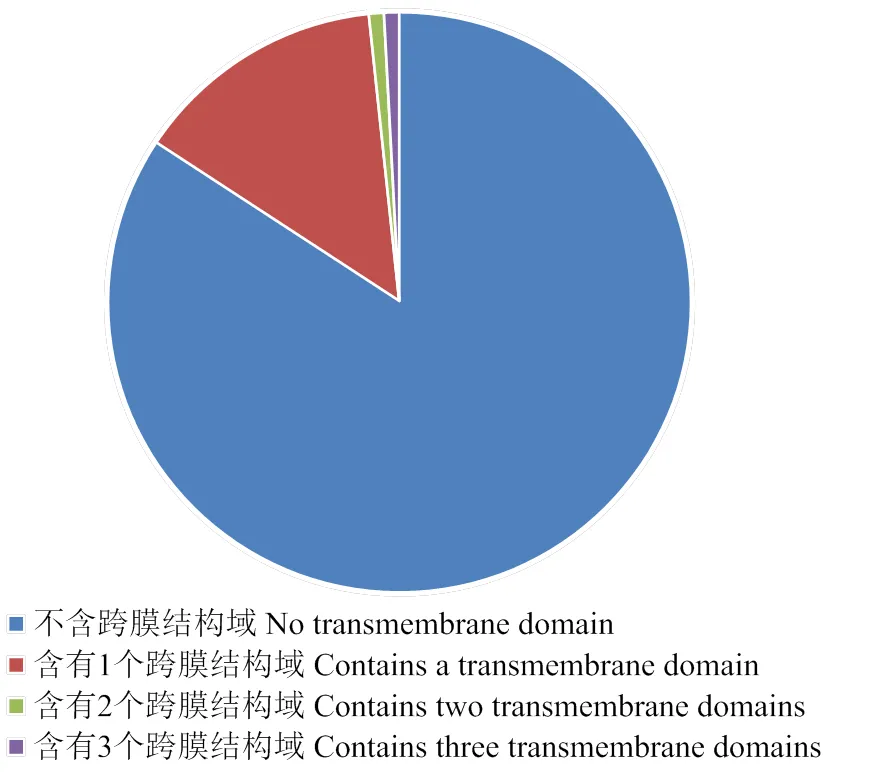
图2 可可毛色二孢中685个外泌蛋白的跨膜结构域分析
为了在预测蛋白中将这一部分蛋白去除,对576个蛋白序列进一步应用big-PI Fungal predictor[26]在线软件进行GPI锚定位点预测分析,结果发现552个蛋白序列不具有GPI锚定位点,而24个蛋白含有GPI锚定位点。通过对以上552个非GPI锚定蛋白序列测试发现,这552个测试蛋白序列均具有胞外分泌途径信号肽(SP)。
综合运用上述5种生物信息学算法程序,对可可毛色二孢全基因组12 902个蛋白序列进行预测筛选,最终获得552个具有信号肽、不含有跨膜结构域和GPI锚定位点并可外泌到细胞外的具有典型特征的经典分泌蛋白,占全基因组预测蛋白总数的4.3%。
2.2 可可毛色二孢中分泌蛋白的信号肽特征
在552个具有分泌蛋白信号肽的蛋白序列中,蛋白序列长度最小的是66 aa,最大的长度是2 269 aa,大多集中于101—400 aa,占蛋白序列总数的54.5%(图3),上述结果表明,可可毛色二孢中所预测的典型分泌蛋白多属于小型蛋白。
信号肽氨基酸长度统计结果如图4所示,候选分泌蛋白的信号肽长度多集中在18—20 aa,有253个,占预测分泌蛋白总数的45.8%;其中,含信号肽长度为20 aa的蛋白数量最多,有91个,占比16.5%。关于候选分泌蛋白的信号肽酶识别位点,利用LipoP v1.0[28]在线程序进行预测分析,结果表明含有Sp I型信号肽识别位点的分泌蛋白序列有522个,占比94.6%;另有10个分泌蛋白含有Sp II型信号肽识别位点,2个含有CYT型信号肽识别位点。以上结果说明大部分的可可毛色二孢候选分泌蛋白是通过Sp I型信号肽酶识别并切割掉信号肽。
进一步对20种氨基酸在候选分泌蛋白信号肽区段中的使用频率进行统计分析,结果如图5所示,在组成信号肽的20种氨基酸中,有129个丙氨酸(A),数量最多,占分泌蛋白总数的23.4%;其次为亮氨酸(L),数量为106个,占全部的19.2%;非极性、疏水的氨基酸使用频率最高(A、G、I、L、P和V),占蛋白总数的60.2%;其次是极性、不带电荷的氨基酸(C、M、N、Q、S和T),占全部的27.2%;带正电荷的碱性氨基酸(K、R和H)所占比例为5.2%;带负电荷的酸性氨基酸(A和E)占23.7%;芳香族氨基酸(W、F和Y)占6.8%。

图3 可可毛色二孢典型分泌蛋白的序列长度

图4 可可毛色二孢分泌蛋白的信号肽长度分布

图5 可可毛色二孢分泌蛋白氨基酸使用频率
对可可毛色二孢候选分泌蛋白信号肽切割位点进行统计分析,结果如表3所示,在信号肽和成熟分泌蛋白切割位点-3、-2、-1、1、2、3位最多的氨基酸分别为A、L、A、A、P、T,所占比例分别为47.0%、16.4%、73.0%、27.3%、27.9%、14.0%。位于信号肽切割位点之前的-3、-2、-1位的氨基酸组成为A-S-A,属于比较典型的A-X-A类型,可被Sp I型信号肽酶识别并切割,这与LipoP v1.0预测的结果相一致。
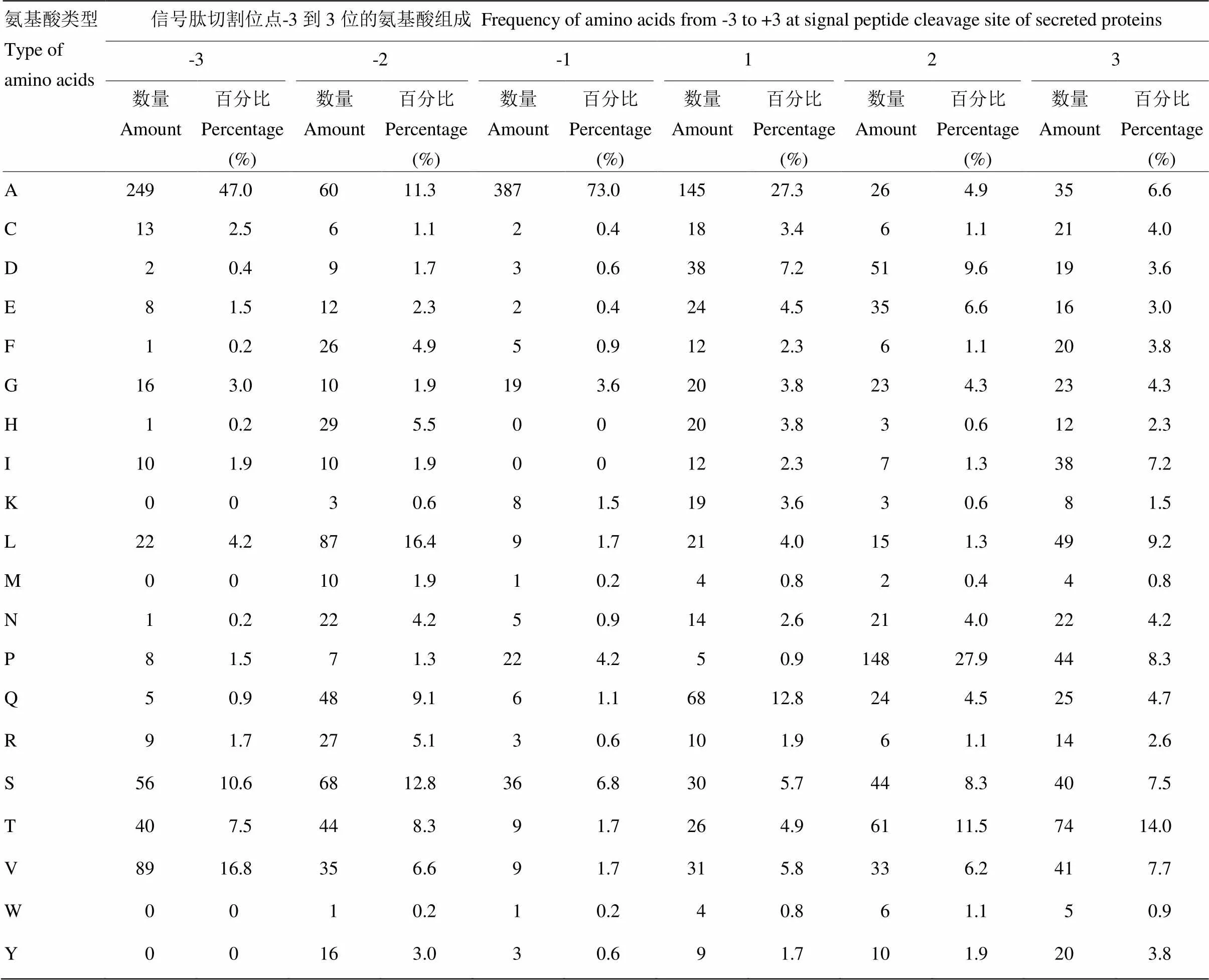
表3 可可毛色二孢分泌蛋白信号肽切割位点的氨基酸组成分布
2.3 可可毛色二孢全基因组候选分泌蛋白的功能预测
将预测的可可毛色二孢候选分泌蛋白在NCBI数据库进行比对分析发现,216个分泌蛋白描述为功能未知的推定蛋白,其余336个则有明确的功能描述。在已有功能描述的这些分泌蛋白中,注释功能主要集中在细胞壁组分降解相关的酶类,如蛋白酶、糖基水解酶、纤维素酶、角质酶、果胶裂解酶等,占比41.7%;此外还有CFEM结构域蛋白、FAD结合结构域蛋白、LysM结构域蛋白等结构域蛋白,以及与致病侵染相关的坏死诱导相关蛋白以及几丁质结合蛋白等(表4)。并且上述候选分泌蛋白在序列长度、等电点、分子量和脂肪族氨基酸指数等方面均存在差异,推测其可能参与不同的生理活动。
2.4 分泌蛋白预测信号肽的生物学功能分析
借助pSUC2系统[30-31]验证候选分泌蛋白的信号肽是否具有分泌功能。SUC2编码一个果糖苷酶,可将蔗糖、棉籽糖等多糖酶解生成葡萄糖、果糖等单糖。将9个候选可可毛色二孢分泌蛋白信号肽融合至SUC2蛋白的氨基端,转至酵母突变体YTK12中,若pSUC2融合载体成功转化到酵母中,则能在CMD-W培养基中正常生长。随后挑去生长的单斑,在棉籽糖培养基(YPRAA)上划线,若预测的分泌蛋白信号肽能够引导SUC2向胞外分泌,则可分泌到酵母细胞外将YPRAA培养基中的棉籽糖分解成单糖,酵母突变体就能正常生长,反之,则不会生长。结果如图6所示,阴性对照Mg87酵母菌不能在YPRAA培养基上存活,阳性对照Avr1b的酵母菌可以在YPRAA培养基生长,而预测的9个分泌蛋白融合载体转化的酵母可在YPRAA培养基上生长,表明上述分泌蛋白信号肽具有分泌活性,是典型的分泌蛋白。
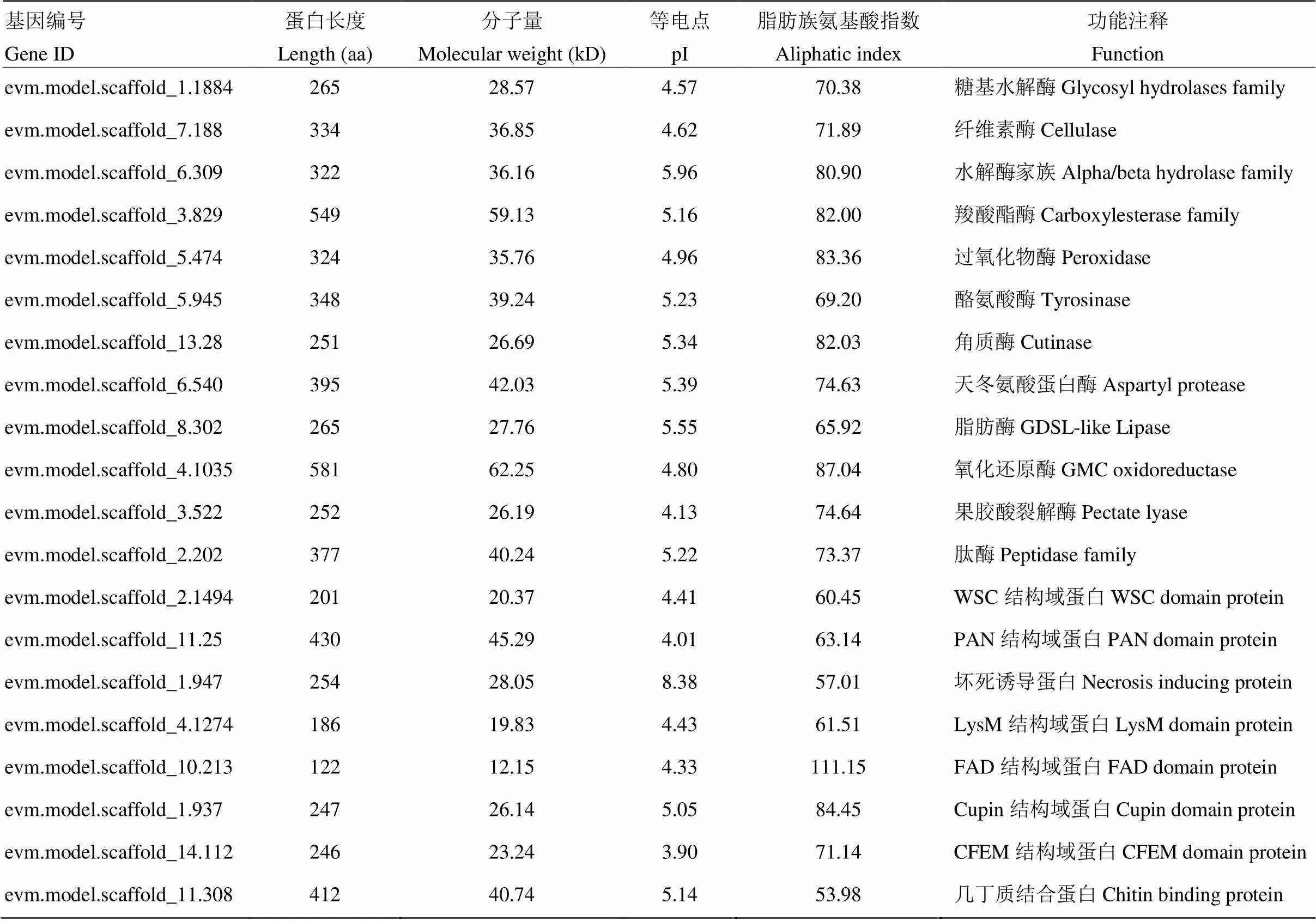
表4 可可毛色二孢部分分泌蛋白生化特性与功能注释
2.5 可可毛色二孢候选分泌蛋白基因的表达分析
在可可毛色二孢侵染初期,和的表达是持续下降的趋势;在接种6 h后的表达量是接种0 h的2.5倍,随后表达下调;、、、和的表达持续上调;而在接种12 h才表现出诱导表达(图7)。结果表明,上述分泌蛋白可能在可可毛色二孢的侵染初期发挥一定的作用。
3 讨论
在与宿主植物的协同进化过程中,病原真菌衍化出多种攻击宿主的策略[11,32]。病原菌会通过分泌大量蛋白质来干扰宿主细胞功能并诱导其免疫反应,从而促进病原菌的侵染[14,33]。因此,利用全基因组测序信息,研究不同病原菌中分泌蛋白的数量、类型和特征以探究其致病机理尤为重要[34-36]。葡萄座腔病菌全基因组测序的完成和公布为该菌激发子、致病蛋白和效应子以及与宿主葡萄的互作研究提供了重要的数据支撑[22]。基于病原真菌分泌蛋白的典型结构特征,周晓罡等利用生物信息学分析软件已对病原真菌以及卵菌等的分泌蛋白进行了预测[17-21]。本文基于以上研究,运用5种生物学软件进行预测分析,保证了测试结果的准确性。LIU等研究表明,在侵染过程中,大豆疫霉()分泌蛋白PsIsc1和大丽轮枝菌()分泌蛋白VdIsc1可作为水解酶分解宿主植物的异分支酸合酶,破坏宿主的水杨酸代谢途径,终止水杨酸介导的免疫反应从而使宿主更为感病[35]。值得关注的是,上述2种分泌蛋白均缺乏分泌信号肽。因此,对真菌分泌蛋白的筛选鉴定,不仅需要通过生物信息学的手段并结合生物学试验进行验证,还需要结合双向电泳(2-DE)和质谱为基础的蛋白质组学等技术进行多维度的挖掘[36]。

图6 分泌蛋白信号肽功能验证

图7 9个候选分泌蛋白基因在可可毛色二孢侵染过程中的相对表达量
基于可可毛色二孢全基因组的12 902个蛋白序列,本研究预测得到552个经典分泌蛋白,占比4.3%。大多数候选分泌蛋白属于小型蛋白(图3),其信号肽长度多集中在18—20 aa(图4),能够被Sp I型信号肽识别并切割,这与前人所报道的有关植物病原真菌、卵菌等分泌蛋白无明显差异[16-21]。分泌蛋白信号肽切割位点-3—+3位使用频率最多是A、L、A、A、P、T(表3),切割位点上氨基酸使用种类较大丽轮枝菌[16]要多。切割位点-3和-1位的氨基酸使用种类最少,分别是16和18种(表3),这也与其他病原菌不同[17-21]。氨基酸使用频率变化最大的在-1位,范围从0(H,组氨酸;I,异亮氨酸)到73%(A,丙氨酸),与大丽轮枝菌、马铃薯晚疫病菌()情况类似[16-21],说明这一切割位点位置的保守性。
对于可可毛色二孢候选分泌蛋白功能注释分析发现,39%预测为功能未知的假定蛋白,说明该菌候选蛋白的特异性,其功能有待于进一步验证分析。植物病原真菌可能最初分泌细胞壁降解酶类来消化细胞壁的阻碍,以促进其侵入[37]。在有功能描述的候选分泌蛋白中(表4),其功能主要集中于降解细胞壁组分的酶类,如纤维素酶、果胶裂解酶和果胶酶等,这与其他病原真菌分泌蛋白的功能相似[17-21]。此外,预测有LysM结构域蛋白、几丁质结合蛋白、坏死诱导蛋白等蛋白(表4),这些蛋白可能参与了可可毛色二孢的致病过程[38-39]。随后,借助于pSUC2系统[30-31]对所选分泌蛋白信号肽进行了功能验证,随机挑选的9个候选蛋白信号肽均正常发挥作用,说明本研究预测方法的精准度较高(图6)。进一步通过qPCR表达分析(图7),验证了候选分泌蛋白基因在可可毛色二孢侵染初期不同的表达模式,推测其在病原菌致病过程中发挥着不同的作用。
4 结论
基于病原真菌分泌蛋白具有的典型特征,利用在线生物信息学程序从可可毛色二孢全基因组中共预测获得552个经典分泌蛋白,多属于小型蛋白。其信号肽氨基酸长度分布广泛,氨基酸组成中非极性、疏水的氨基酸使用频率最高。功能注释主要集中在细胞壁组分降解相关的酶类、与致病侵染相关的坏死诱导相关蛋白以及几丁质结合蛋白等。
[1] BERTSH C, LARIGNONP, FARINES, CLEMENT C, FONTAINE F. The spread of grapevine trunk disease., 2009, 324(5928): 721.
[2] YANJY, XIEY, YAOS W, WANG Z Y, LI X H. Characterization of, the causal agent of grapevine canker in China., 2012, 41(4): 351-357.
[3] YAN J Y, XIE Y, ZHANG W, WANG Y, LIU J K, HYDE K D, SEEM R C, ZHANG G Z, WANG Z Y, Yao S W, BAI X J, Dissanayake A J, Peng Y L, Li X H. Species of Botryosphaeriaceae involved in grapevine dieback in China., 2013, 61(1): 221-236.
[4] DISSANAYAKE A J, ZHANG W, LIU M, CHUKEATIROTE E, YAN J Y, LI X H, HYDE K D.causes pedicel and peduncle discolouration of grapes in China., 2015, 10: 21.
[5] DISSANAYAKE A J, ZHANG W, LI X H, ZHOU Y, CHETHANA T, CHUKEATIROTE E, HYDE K D, YAN J Y, ZHANG G Z, ZHAO W S. First report ofassociated with grapevine dieback in China., 2015, 54(2): 414-419.
[6] NIMCHUK Z, EULGEM T, HOLT III B F, DANGL J L. Recognition and response in the plant immune system., 2003, 37: 579-609.
[7] RODRIGUEZ-MORENO L, EBERT M K, BOLTON M D, THOMMA B P H J. Tools of the crook-infection strategies of fungal plant pathogens., 2018, 93(4): 664-674.
[8] GREENBAUM D, LUSCOMBE N M, JANSEN R, QIAN J, GERSTEIN M. Interrelating different types of genomic data, from proteome to secretome: ′oming in on function., 2001, 11(9): 1463-1468.
[9] DE SAIN M, REP M. The role of pathogen-secreted proteins in fungal vascular wilt diseases., 2015, 16(10): 23970-23993.
[10] CHOI J, PARK J, KIM D, JUNG K, KANG S, LEE Y H. Fungal secretome database: integrated platform for annotation of fungal secretomes., 2010, 11: 105.
[11] VAN DER BURGH A M, JOOSTEN M H. Plant immunity: thinking outside and inside the box., 2019,24(7): 587-601.
[12] JONES J D, DANGL J L. The plant immune system., 2006, 444: 323-329.
[13] OLIVEIRA-GARCIA E, VALENT B. How eukaryotic filamentous pathogens evade plant recognition., 2015, 26: 92-101.
[14] ASAI S, SHIRASU K. Plant cells under siege: plant immune system versus pathogen effectors., 2015, 28: 1-8.
[15] FRANCESCHETTI M, MAQBOOL A, JIMENEZ-DALMARONI M J, PENNINGTON H G, KAMOUN S, BANFIELD M J. Effectors of filamentous plant pathogens: commonalities amid diversity., 2017, 81(2): e00066-16.
[16] 田李, 陈捷胤, 陈相永, 汪佳妮, 戴小枫. 大丽轮枝菌(VdLs. 17) 分泌组预测及分析. 中国农业科学, 2011, 44(15): 3142-3153.
TIAN L, CHEN J Y, CHEN X Y, WANG J N, DAI X F. Prediction and analysis ofVdLs. 17 secretome., 2011, 44(15): 3142-3153. (in Chinese)
[17] 周晓罡, 侯思名, 陈铎文, 陶南, 丁玉梅, 孙茂林, 张绍松. 马铃薯晚疫病菌全基因组分泌蛋白的初步分析. 遗传, 2011, 33(7): 785-793.
ZHOU X G, HOU S M, CHEN D W, TAO N, DING Y M, SUN M L, ZHANG S S. Genome-wide analysis of the secreted proteins of., 2011, 33(7): 785-793. (in Chinese)
[18] 陈琦光, 王陈骄子, 杨媚, 周而勋. 希金斯刺盘孢全基因组候选效应分子的预测. 热带作物学报, 2015, 36(6): 1105-1111.
CHEN Q G, WANG C j z, YANG M, ZHOU E X. Prediction of candidate effectors from the genome of, 2015, 36(6): 1105-1111. (in Chinese)
[19] 韩长志. 全基因组预测禾谷炭疽菌的分泌蛋白. 生物技术, 2014, 24(2): 36-41.
Han C Z. Prediction for secreted proteins fromgenome., 2014, 24(2): 36-41. (in Chinese)
[20] DE CARVALHO M C, NASCIMENTO L C, DARBEN L M, POLIZEL- PODANOSQUI A M, LOPES-CAITAR V S, QI M, ROCHA C S, CARAZZOLLE M F, KUWAHARA M K, PEREIRA G A, ABDELNOOR R V, WHITHAM S A, MARCELINO-GUIMARAES F C. Prediction of the in plantasecretome and potential effector families., 2017, 18(3): 363-377.
[21] ZENG R, GAO S G, XU L X, LIU X, DAI F M. Prediction of pathogenesis-related secreted proteins from., 2018, 18(1): 191.
[22] YAN J Y, ZHAO W S, CHEN Z, XING Q K, ZHANG W, CHETHANA K W T, XUE M F, XU J P, PHILLIPS A J L, WANG Y,. Comparative genome and transcriptome analyses reveal adaptations to opportunistic infections in woody plant degrading pathogens of Botryosphaeriaceae., 2018, 25(1): 87-102.
[23] ARMENTEROS J J A, TSIRIGOS K D, SONDERBY C K, PETERSEN T N, WINTHER O, BRUNAK S, VON HEIJNE G, NIELSEN H. SignalP 5.0 improves signal peptide predictions using deep neural networks., 2019, 37(4): 420-423.
[24] EMANUELSSON O, NIELSEN H, BRUNAK S, VON HEIJNE G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence., 2000, 300(4): 1005-1016.
[25] KROGH A, LARSSON B E, VON HEIJNE G, Sonnhammer E L L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes., 2001, 305(3): 567-580.
[26] EISENHABER B, BORK P, EISENHABER F. Post-translational GPI lipid anchor modification of proteins in kingdoms of life: analysis of protein sequence data from complete genomes., 2001, 14(1): 17-25.
[27] ARMENTEROS J J A, SALVATORE M, EMANUELSSON O, WINTHER O, VON HEIJNE G, ELOFSSON A, NIELSEN H. Detecting sequence signals in targeting peptides using deep learning., 2019, 2(5): e201900429.
[28] JUNCKER A S, WILLENBROCK H, VON HEIJNE G, Nielsen H, Brunak S, Krogh A. Prediction of lipoprotein signal peptides in Gram-negative bacteria., 2003, 12(8): 1652-1662.
[29] GASTEIGER E, HOOGLAND C, GATTIKER A, DUVAUD S, WILKINS M R, APPEL R D, BAIROCH A. Protein identification and analysis tools on the ExPASy server//. Humana Press, 2005: 571-607.
[30] JACOBS K A, COLLINS-RACIE L A, COLBERT M, DUCKETT M, GOLDEN-FLEET M, KELLEHER K, KRIZ R, LAVALLIE E R, MERBERG D, SPAULDING V, STOVER J, WILLIAMSON M J, MCCOY J M. A genetic selection for isolating cDNAs encoding secreted proteins., 1997, 198(1/2): 289-296.
[31] FANG A F, HAN Y Q, ZHANG N, ZHANG M, LIU L J, LI S, LU F, SUN W X. Identification and characterization of plant cell death- inducing secreted proteins from., 2016, 29(5): 405-416.
[32] SONAH H, DESHMUKH R K, BELANGER R R. Computational prediction of effector proteins in fungi: opportunities and challenges., 2016, 7: 126.
[33] WAN W L, FROHLICH K, PRUITT R N, NURNBERGER T, Zhang L. Plant cell surface immune receptor complex signaling., 2019, 50: 18-28.
[34] TORUNO T Y, STERGIOPOULOS I, COAKER G. Plant-pathogen effectors: cellular probes interfering with plant defenses in spatial and temporal manners., 2016, 54: 419-441.
[35] LIU T, SONG T, ZHANG X, YUAN H, SU L, LI W, XU J, LIU S, CHEN L, CHEN T, ZHANG M, Gu L, ZHANG B, DOU D. Unconventionally secreted effectors of two filamentous pathogens target plant salicylate biosynthesis., 2014, 5: 4686.
[36] 李云锋, 聂燕芳, 王振中. 植物病原真菌分泌蛋白质组学研究进展. 微生物学通报, 2015, 42(6): 1101-1107.
LI Y F, NIE Y F, WANG Z Z. Research progress on secretomics of phytopathogenic fungi., 2015, 42(6): 1101-1107. (in Chinese)
[37] BROUWER H, COUTINHO P M, HENRISSAT B, DE VRIES R P. Carbohydrate-related enzymes of important Phytophthora plant pathogens., 2014, 72: 192-200.
[38] SANCHEZ-Vallet A, MESTERS J R, THOMMA B P. The battle for chitin recognition in plant-microbe interactions., 2015, 39(2): 171-183.
[39] AKCAPINAR G B, KAPPEL L, SEZERMAN O U, SEIDL-SEIbothV. Molecular diversity of LysM carbohydrate-binding motifs in fungi., 2015, 61(2): 103-113.
Prediction and analysis of Candidate Secreted Proteins from the Genome of
XING QiKai1, LI LingXian1, Cao Yang2, ZHANG Wei1, PENG JunBo1, YAN JiYe1, LI XingHong1
(1Institute of Plant and Environment Protection, Beijing Academy of Agriculture and Forestry Sciences/Beijing Key Laboratory of Environment Friendly Management on Fruit Diseases and Pests in North China, Beijing 100097;2School of Biological Engineering, Dalian University of Technology, Dalian 116024, Liaoning)
【】is an important phytopathogenic fungus with a worldwide distribution. This species causes severe Botryosphaeria dieback on a wide range of woody plants, which leads to reduced crop quality and tremendous economic losses. The objective of this study is to predict and analyze the candidate secreted proteins in the genome of, clarify their basic characteristics, so as to lay a foundation for the study of the pathogenic mechanism of secreted proteins in this pathogen.【】The signal peptide prediction algorithm SignalP v5.0 and subcellular localization prediction algorithm ProtComp v9.0, transmembrane helix prediction algorithm TMHMM v2.0, GPI-anchoring site prediction algorithm big-PI Fungal Predictor, and subcellular protein location distribution algorithm TargetP v2.0 were used to analyze 12 902 protein sequences ofpublished in the previous study. The basic features including the length of the N-terminal signal peptide, the frequency of amino acid usage and cleavage site of the predicted secreted proteins were statistically analyzed. Based on the homology of the protein sequence, biological function annotation of the predicted secreted proteins was clarified by using BLASTP program. The activity of the signal peptide of the selected secreted proteins was detected by yeast secretion and cell translocation assays. Expression patterns of the selected secreted protein genes duringinfection were analyzed by qRT-PCR technology.【】In this study, 522 secreted proteins were verified, accounting for 4.3% of the total proteins present in the genome of. The lengths of amino acids of secreted proteins were ranged from 101 to 400 aa. The distribution length of signal peptides was from 18 to 20 aa and the largest number was 20 aa. The top frequent amino acid was alanine in the signal peptides, and the most frequently incorporated amino acids were non-polar and hydrophobic, accounting for 60.2% of the total amino acids. Further, the amino acids in the position -3 to -1 in the signal peptides were relatively conserved and the signal peptide cleavage site belonged to A-X-A type, which could be recognized and cleaved by Sp I type peptidase. Among them, 336 secreted proteins were identified with a predictive function, which was mostly enzymatic or virulence-associated protein. Besides, there are differences in terms of molecular weight, isoelectric point, the aliphatic index in the candidate secreted proteins.Finally, the predicted signal peptides of the 9 putativesecreted proteins were confirmed to have secretory activity by using a yeast invertase secretion assay. qRT-PCR analysis demonstrated that the expression of selected protein genes was differentially regulated during host infection.【】A total of 552 candidate secreted proteins ofwere predicted by a set of computer algorithms. Lengths of the signal peptides vary greatly and the most frequently are mainly non-polar and hydrophobic amino acids. Secreted proteins characterized in this study can be categorized under enzymes related to the degradation of cell wall components, necrosis induction proteins, and chitin-binding proteins which may play an important role inpathogenetic mechanism.
Botryosphaeria dieback;; bioinformatics; secreted protein; signal peptide; expression pattern

10.3864/j.issn.0578-1752.2020.24.006
2020-02-14;
2020-03-20
国家自然科学基金(31801686)、北京市自然科学基金(6184041)、北京市农林科学院科技创新能力建设(KJCX20190406)
邢启凯,E-mail:qikaixing@163.com。通信作者李兴红,Tel:010-51503510;E-mail:lixinghong1962@163.com
(责任编辑 岳梅)
