宜昌腊肠与恩施腊肠细菌多样性的比较分析
2020-12-28周亚澳黄怡王韵博郭壮张振东李杨坤侯强川
周亚澳 黄怡 王韵博 郭壮 张振东 李杨坤 侯强川


摘 要:采用Illumina MiSeq高通量测序技术对宜昌腊肠细菌多样性进行解析,并与之前报道的恩施腊肠细菌群落结构进行比较分析。结果表明:宜昌腊肠在门水平主要由厚壁菌门(Firmicutes,64.93%)、变形菌门(Proteobacteria,32.12%)和放线菌门(Actinobacteria,1.14%)组成;在属水平上,主要由葡萄球菌属(Staphylococcus,16.57%)、乳酸杆菌属(Lactobacillus,16.22%)、魏斯氏菌属(Weissella,14.67%)和明串珠菌属(Leuconostoc,11.43%)等组成。宜昌腊肠細菌丰度显著低于恩施腊肠(P<0.05),且2 个地区腊肠的细菌菌群结构存在较大差异。基因功能预测结果表明,宜昌腊肠中细菌在碳水化合物代谢和多糖的生物合成与代谢等方面更加旺盛,这些差异与腊肠中乳酸杆菌的相对丰度存在显著相关性。
关键词:腊肠;高通量测序;微生物群落结构;功能预测
Abstract: The bacterial diversity in Yichang sausage was analyzed by Illumina MiSeq high-throughput sequencing technique and compared with that previously reported for Enshi sausage. The results showed that the bacterial community in Yichang sausages were mainly composed of Firmicutes (64.93%), Proteobacteria (32.12%) and Actinobacteria (1.14%) at the phylum level. At the genus level, it was mainly composed of Staphylococcus (16.57%), Lactobacillus (16.22%), Weissella (14.67%), and Leuconostoc (11.43%). The abundance of bacteria in Yichang sausages was significantly lower than that in Enshi sausage (P < 0.05). Besides, there were great differences in the bacterial structure between the two sausages. The results of gene function prediction showed that bacteria in Yichang sausages were more vigorous in carbohydrate metabolism, and polysaccharide biosynthesis and metabolism, and these differences were significantly correlated with the relative abundance of Lactobacillus in sausages.
Keywords: sausages; high throughput sequencing; microbial community structure; function prediction
DOI:10.7506/rlyj1001-8123-20200711-169
中图分类号:TS251.5 文献标志码:A 文章编号:1001-8123(2020)10-0008-06
腊肠作为我国一种传统的发酵肉制品,拥有悠久的制作历史,现广泛分布于我国华东、华中和华南等地区[1]。腊肠的制作主要以猪肉为原料,同时辅以食盐、八角、花椒和白酒等调味料进行腌制,灌入肠衣后进行晾晒发酵[2]。因其风味独特、口感醇厚而深受广大消费者的喜爱[3]。
宜昌市位于湖北省西南部,地处长江上游与中游结合部,其气候为腊肠的发酵提供了有利条件。同时宜昌市是一个多民族杂居的城市,这在一定程度促进了传统腊肠制作工艺的融合发展。传统腊肠的制作环境较为粗犷,不同的地理环境和气候条件可能会赋予腊肠不同的微生物群系[4]。目前,一些学者针对不同品质、原料及发酵和贮藏阶段腊肠微生物群落组成和差异开展一些研究,滕安国等[5]发现乳杆菌属(Lactobacillus)和明串珠菌属(Leuconostoc)为正常腊肠和肠衣中的优势菌属,而腊肠发黏变质后微生物组成发生变化,菌群多样性降低;刘长建等[6]从腊肠中分离得到多株具有降胆固醇能力的乳酸菌,其中干酪乳杆菌(Lactobacillus casei)的降胆固醇效果最好;田甜等[7]发现在广式腊肠发酵过程中添加复合发酵剂能加快发酵进程,并降低亚硝酸盐含量。Fougy等[8]发现减少腊肠中的食盐添加量会导致腐败菌生长和细菌多样性下降;Li Xinfu等[9]通过高通量测序技术解析真空包装腊肠贮藏过程中微生物群落的变化。此外,越来越多的研究表明,腊肠的发酵菌群直接影响着发酵的进程和产品品质,郭壮等[10]研究发现腊肠中的乳酸菌对产品发酵成熟和风味品质的形成具有至关重要的作用;黄金枝[11]研究发酵菌种与广式腊肠产品品质的关系,发现戊糖片球菌(Pediococcus pentosaceus)对广式腊肠发酵品质的影响大于汉逊德巴利酵母(Saccharomyces)和植物乳杆菌(Lactobacillus plantarum)。上述研究的结果表明,腊肠品质与其细菌组成之间存在密切联系。然而,目前鲜有研究报道宜昌地区腊肠细菌多样性及其与其他地区腊肠菌群结构的差异,这在一定程度上限制了宜昌地区腊肠品质的提升。
近年来,伴随分子生物学的发展,越来越多的基于非培养的微生物检测方法和技术被广泛应用于发酵食品微生物检测中。其中,Illumina MiSeq高通量测序技术因其检测速率快、通量大和结果可信度高等优点[12-13],在海洋、土壤、发酵食品和肠道等环境样品微生物多样性的研究中得到广泛应用[14-15]。邓风等[16]采用聚合酶链式反应(polymerase chain reaction,PCR)-变性梯度凝胶电泳与Illumina MiSeq高通量测序技术相结合的方法研究恩施地区腊肠中的细菌群落结构,发现恩施腊肠样品中含有大量的共有细菌类群,優势细菌属主要包括环丝菌属(Brochothrix)、葡萄球菌(Staphylococcus)、嗜冷杆菌(Psychrobacter)等,同时恩施腊肠所有测序序列中无法鉴定到属水平的仅为13.72%。该研究结果进一步表明Illumina MiSeq高通量测序可以作为解析腊肠细菌群落结构的有效技术手段。
本研究采用Illumina MiSeq高通量测序技术对采集自宜昌地区的腊肠细菌多样性进行解析。此外,通过整合恩施腊肠测序数据[17],结合多元统计学分析和功能预测等手段比较分析2 个地区腊肠细菌菌群结构和功能的差异。以期进一步丰富人们对不同地区腊肠细菌群落结构的认识。
1 材料与方法
1.1 材料与试剂
腊肠采集自湖北省宜昌市五峰土家族自治县菜市场,共计9 份,编号记为YC1~YC9,所有样品均为手工制作,且满足以下标准:1)腊肠产品在宜昌地区生产制作完成;2)腊肠使用猪肉作为制作原料;3)腊肠产品无霉变、无异味且无明显杂质存在。样品采集时使用无菌手术刀切取约100 g装入无菌采样袋,密封后置于采样箱中,低温冷藏运回实验室进行后续操作。
338F/806R引物 武汉天一辉远生物科技有限公司;DNeasy Mericon Food Kit 德国Qiagen公司;FastPfu Fly DNA Polymerase、5×TransStart? FastPfu缓冲液和dNTPs Mix 北京全式金生物技术有限公司。
1.2 仪器与设备
Vetiri梯度基因扩增仪 美国AB SCIEX公司;ND-2000C微量紫外分光光度计 美国Nano Drop公司;Illumina MiSeq PE300高通量测序平台 美国Illumina公司;R960机架式服务器 美国Dell公司;UVPCDS8000凝胶成像分析系统、DCode? System 美国Bio-Rad公司;CR21N型高速离心机 日本日立金属株式会社。
1.3 方法
1.3.1 样品预处理及微生物宏基因组DNA提取
将采集的腊肠切碎后,称取5 g加入到45 mL生理盐水中,使用拍击器拍击5 min,400 r/min离心8 min后取上清液,于12 000 r/min离心8 min,取沉淀备用[16]。使用基因组提取试剂盒提取沉淀物宏基因组DNA。
1.3.2 PCR扩增和Illumina MiSeq测序
使用338F/806R引物(带有7 个碱基核苷酸标签)对细菌16S rRNA V3~V4区进行PCR扩增,扩增条件参照王玉荣等[17]方法。使用1.00%的琼脂糖凝胶电泳和紫外分光光度计对扩增产物的纯度和浓度进行检验,将合格DNA产物送至上海美吉生物医药科技有限公司进行测序。
1.3.3 生物信息学分析
依照郭壮等[18]研究中序列质控所约束的参数对下机序列进行质量控制,并依照核苷酸标签进行序列的分配,将质控后的序列分配到不同的样本中。参照Yang Chengcong等[19]的方法,使用QIIME V1.9.1平台对宜昌和恩施腊肠中的细菌组成和多样性进行解析。具体的生物学分析过程如下:1)采用PyNAST软件对序列数据进行比对和校准[20];2)使用UCLUST分别通过100%和97%相似度对校准后的序列进行划分[21],分别得到非冗余数据集和操作分类单元(operational taxonomic units,OTU);3)使用ChimeraSlayer软件识别OTU中的嵌合体序列[22],并对相应的嵌合体序列进行删除;4)挑选每个OTU中的代表性序列分别使用Greengene(Version 13.8)[23]、RDP(Relace 11.5)[24]和Sliva(Version 132)数据库[25]进行同源性比对,确定各OTU的分类学地位,使用内部脚本整合3 个数据库的注释结果,确定各OTU的最终分类学地位;5)使用FastTree软件构建基于OTU代表性序列的系统发育树[26];6)依据最大序列数计算每个腊肠样本的α多样性指数(Chao 1指数、Shannon指数、Observed species指数和Simpson指数);7)依据最小序列数计算样品间的β多样性。
使用PICRUSt(Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)软件对腊肠中细菌微生物的基因功能进行预测[27],并参照京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)数据库进行基因功能注释。
1.3.4 多元统计学分析
使用非参数的Mann-Whitney秩和检验对不同地区腊肠样品菌群组成进行差异显著性分析;使用LefSe软件分析并基于LDA Effect Size判别组间丰度有显著差异的微生物;基于Pearson相关性分析腊肠中优势菌属和差异代谢通路之间的相关性。使用R软件(v3.5.0)、STAMP软件和Origin 2019b软件进行数据可视化处理。
2 结果与分析
2.1 宜昌腊肠序列丰富度和多样性分析
宜昌腊肠所有样品共测得序列427 570 条,每个样品的平均测序量为26 009 条(12 215~40 828,标准偏差3 192),上述序列在97%相似度划分得到5 330 个非嵌合体OTU,每个样本平均OUT数为931。
由表1可知,YC9腊肠的OTU数和Chao 1指数最大,而YC2腊肠的Shannon指数最大。通过对OTU数、Chao 1指数和Shannon指数的变异系数进行计算发现,宜昌腊肠在α多样性指数上存在较大的组内差异。相关研究显示,Chao 1指数和Shannon指数常分别用来评估环境中微生物的丰度和多样性[28]。表明YC9腊肠中微生物丰度最高,而YC2腊肠的微生物多样性最高,且宜昌腊肠中的微生物丰度和多样性存在较大差异。
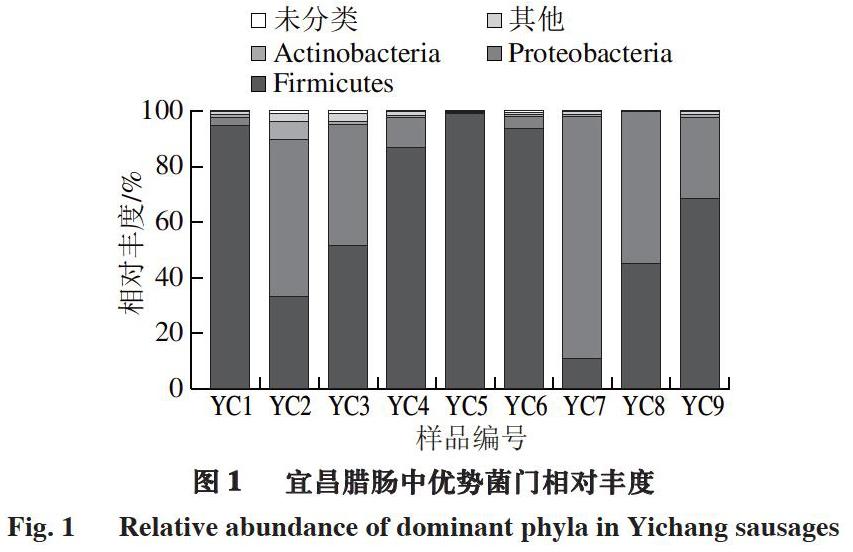
进一步分析宜昌腊肠中的细菌菌落组成,所有样品共注释得到15 个菌门。由图1可知,宜昌腊肠中优势菌门(平均相对丰度大于1.00%)共有3 个,分别为厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)和放线菌门(Actinobacteria),平均相对丰度分别为64.93%、32.12%和1.14%。可见,宜昌腊肠中的主要菌门为厚壁菌门和变形菌门,这与邓风等[16]研究得到的恩施腊肠细菌群落结构结果基本一致。
本研究进一步在属水平对宜昌腊肠细菌群落结构进行分析,由图2可知,宜昌腊肠中优势菌属(平均相对丰度大于1.00%)共有14 个,其中葡萄球菌属(Staphylococcus,16.57%)、乳酸杆菌属(Lactobacillus,16.22%)、魏斯氏菌属(Weissella,14.67%)、明串珠菌属(Leuconostoc,11.43%)和环丝菌属(Brochothrix,2.54%)隶属于厚壁菌门;拉恩菌属(Rahnella,7.91%)、假单胞菌属(Pseudomonas,5.74%)、泛菌属(Pantoea,3.16%)、克吕沃尔氏菌属(Kluyvera,2.76%)、肠杆菌属(Enterobacter,2.21%)、乳球菌属(Lactococcus,1.67%)、柠檬酸杆菌属(Citrobacter,1.43%)、拉乌尔菌属(Raoultella,1.21%)和沙雷菌属(Serratia,1.14%)隶属于变形菌门。可见,宜昌腊肠中相对丰度最高的是葡萄球菌属和乳酸杆菌属,这与恩施腊肠中的优势菌属不尽相同,后者优势菌属除葡萄球菌属和乳酸杆菌属外,还包括环丝菌属,三者在样品中的平均相对丰度分别为9.79%、2.80%和38.34%[16]。宜昌腊肠中细菌主要以乳酸菌为主,其平均相对丰度接近60%(主要以乳酸杆菌属、魏斯氏菌属和明串珠菌属为主)。乳酸菌作为发酵肉制品中常见的发酵菌种,在一定程度上决定着发酵制品最终品质。值得注意的是,本研究在宜昌腊肠中检测到大量的机会性致病微生物如葡萄球菌属和沙雷菌属等,提示需要进一步规范化腊肠制作过程的安全管理和质量控制。
2.2 宜昌和恩施腊肠中细菌群落结构的比较分析
由图3可知,宜昌腊肠中细菌的Chao 1指数极显著低于恩施腊肠(P<0.01),Observed species指数显著低于恩施腊肠(P<0.05),Shannon指数低于恩施腊肠,且差异不显著,而Simpson指数略高于恩施腊肠。Chao 1指数和Observed species指数常用于评估环境样本中微生物的丰度,Shannon指数和Simpson指数常用于评估环境中微生物的多样性[28]。可见,宜昌腊肠中微生物的丰度明显低于恩施腊肠,而多样性没有明显差异。
本研究进一步通过基于非加权和加权UniFrac距离的主坐标分析比较2 个地区腊肠菌群的组间差异。由基于非加权UniFrac距离的主坐标分析结果(图4A)可知,宜昌和恩施腊肠的微生物菌群在空间排布上呈现明显的分离趋势,宜昌腊肠和恩施腊肠分别位于X轴的正方向和负方向,多元方差分析结果也表明二者的微生物群落结构差异显著(P<0.05);而基于加权UniFrac距离的分析结果(图4B)表明,虽然2 个地区的腊肠样品整体存在分离趋势,但部分样品在空间排布上出现一定的重叠。上述结果表明,宜昌和恩施腊肠中优势菌群组成较为相似,但二者均含有较多独特的低丰度菌群,这部分菌群对腊肠的整体菌群结构产生较大影响。
為揭示宜昌和恩施腊肠的菌群结构差异,通过LefSe分析比较两地区腊肠存在显著差异的菌群。由图5可知,共有7 个细菌类群(判别得分>3)在2 组腊肠样品中存在显著差异(P<0.05),其主要为果胶杆菌属(Pectobacterium)、肠杆菌属和假单胞菌属,值得注意的是,甄别出的所有差异菌均在恩施腊肠中的相对丰度显著较高(P<0.05)。相关报道显示,肠杆菌属和假单胞菌属广泛存在于传统发酵食品中,能够有效促进发酵制品的发酵进程,其中假单胞菌属与发酵制品的颜色密切相关,且对酪胺和组胺的产生具有一定的影响[29]。同时,发酵制品中的菌群结构与地理环境、制作工艺和原材料等有直接的关系,不同菌群结构可能是造成不同地区腊肠色泽、滋味和风味差异的主要原因之一。通过对不同地区发酵制品中微生物多样性进行解析和菌株的收集及保藏,将对传统发酵食品的工业化和个性化生产具有重要意思。
2.3 PICRUSt功能预测
本研究利用PICRUSt软件对宜昌和恩施腊肠中细菌类群的基因功能进行预测,使用KEGG数据库对预测基因进行功能注释,在此基础上对差异代谢通路进行分析。Z值是用于衡量样本均值偏离整体均值的方差倍数,当用于差异代谢通路查找时,Z值越大,则相应的代谢通路在两组间的差异越大。本研究选择Z值>1.6(95%置信区间)[30],同时代谢通路中的基因在某一地区样品高表达概率>80%作为判定2 个地区差异代谢通路的条件。
由图6A可知,宜昌和恩施腊肠中的细菌菌群在4 条代谢通路上存在显著差异,且所有差异代谢通路均隶属于与代谢相关的一级功能层。其中与碳水化合物和多糖生物合成及代谢的相关基因在宜昌腊肠微生物中显著富集,而与能量代谢和氨基酸代谢的相关基因在恩施腊肠微生物中显著富集。由图6B可知,魏斯氏菌属与氨基酸代谢呈显著负相关(P<0.05),而乳酸杆菌属与多糖生物合成及代谢呈显著正相关(P<0.05),与碳水化合物代谢呈极显著正相关(P<0.01)。可见,宜昌地区腊肠中微生物对碳水化合物的代谢更为旺盛,并且这与腊肠中的乳酸杆菌密切相关。乳酸杆菌属对碳水化合物的利用效率受到环境的温度、湿度等因素的强烈影响[31]。宜昌地区相较恩施地区空气湿度更低,且平均温度更高,上述环境因素均有利于腊肠中乳酸杆菌的生长和代谢,这可能是宜昌腊肠中微生物碳水化合物代谢相关基因富集的原因之一。亮氨酸和异亮氨酸均为人体的必需氨基酸,由于人体无法合成,因此必须从饮食中摄取。恩施腊肠中微生物亮氨酸和异亮氨酸生物合成通路更为活跃,提示该地区生产的腊肠中这2 种必需氨基酸的含量可能会更为丰富。
3 结 论
本研究采用Illumina MiSeq高通量测序技术对湖北宜昌腊肠中的细菌多样性进行解析,并和恩施腊肠进行比较分析。结果表明,宜昌腊肠中的细菌主要隶属于厚壁菌门、变形菌门和放线菌门,总相对丰度98.19%;而优势菌属主要以葡萄球菌属、乳酸杆菌属、魏斯氏菌属和明串珠菌属等为主。宜昌腊肠中的微生物丰度显著低于恩施腊肠,且果胶杆菌属、肠杆菌属和假单胞菌属等菌属在宜昌地区的相对丰度显著低于恩施地区。功能预测结果表明,宜昌和恩施地区腊肠与代谢相关的通路存在差异显著,宜昌腊肠中细菌碳水化合物代谢和多糖生物合成与代谢更加旺盛,这些差异可能与2 个地区的气候不同存在一定关系。
参考文献:
[1] 龙强, 聂乾忠, 刘成国. 发酵肉制品功能性发酵剂研究现状[J]. 食品科学, 2016, 37(17): 263-269. DOI:10.7506/spkx1002-6630-201617044.
[2] 杨江, 杨成聪, 凌霞, 等. 基于电子鼻和气相色谱-质谱联用技术评价襄阳地区腊肠风味品质[J]. 肉类研究, 2018, 32(8): 46-50. DOI:10.7506/rlyj1001-8123-201808008.
[3] 刘琨毅, 王琪, 王卫, 等. 茶多酚对低盐中式腊肠防腐保鲜的影响[J]. 肉类研究, 2018, 32(3): 34-39. DOI:10.7506/rlyj1001-8123-201803007.
[4] 杜莎, 李柯, 黄晴, 等. 人工发酵剂对湘西腊肠微生物菌群和理化特性的影响[J]. 现代食品科技, 2017, 33(4): 241-247. DOI:10.13982/j.mfst.1673-9078.2017.4.037.
[5] 滕安国, 齐晓娜, 张芹, 等. 高通量测序技术分析天然肠衣香肠的菌群组成[J]. 食品研究与开发, 2016, 37(11): 113-116. DOI:10.3969/j.issn.1005-6521.2016.11.027.
[6] 刘长建, 蒋本国, 姜波, 等. 腊肠中降胆固醇乳酸菌的筛选及鉴定[J]. 中国酿造, 2011, 30(8): 35-37. DOI:10.3969/j.issn.0254-5071.2011.08.011.
[7] 田甜, 詹锐琪, 张雅琳, 等. 微生物发酵技术对广味香肠安全性提升的研究[J]. 中国调味品, 2019, 44(12): 26-30. DOI:10.3969/j.issn.1000-9973.2019.12.006.
[8] FOUGY L, DESMONTS M, COEURET G, et al. Reducing salt in raw pork sausages increases spoilage and correlates with reduced bacterial diversity[J]. Applied and Environmental Microbiology, 2016, 82(13): 3928-3939. DOI:10.1128/aem.00323-16.
[9] LI Xinfu, LI Cong, YE Hua, et al. Changes in the microbial communities in vacuum-packaged smoked bacon during storage[J]. Food Microbiology, 2019, 77(2): 26-37. DOI:10.1016/j.fm.2018.08.007.
[10] 郭壯, 王玉荣, 葛东颖, 等. 腊肠发酵过程中细菌多样性评价及其对风味的影响[J].食品科学, 2020. DOI:10.7506/spkx1002-6630-20191121-255.
[11] 黄金枝. 发酵广式腊肠加工菌种优选及品质变化研究[D]. 南昌: 江西农业大学, 2015.
[12] LIANG Huipeng, YIN Liguo, ZHANG Yahao, et al. Dynamics and diversity of a microbial community during the fermentation of industrialized Qingcai paocai, a traditional Chinese fermented vegetable food, as assessed by Illumina MiSeq sequencing, DGGE and qPCR assay[J]. Annals of Microbiology, 2018, 68(2): 111-122. DOI:10.1007/s13213-017-1321-z.
[13] SCHIRMER M, IJAZ U Z, DAMORE R, et al. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform[J]. Nucleic Acids Research, 2015, 43(6): e37. DOI:10.1093/nar/gku1341.
[14] WALSH A M, FIONA C, KIERAN K, et al. Microbial succession and flavor production in the fermented dairy beverage kefir[J]. mSystems, 2016, 1(5): 2379-5077. DOI:10.1128/mSystems.00052-16.
[15] HE Guoqing, LIU Tongjie, ASADIQ F, et al. Insights into the microbial diversity and community dynamics of Chinese traditional fermented foods from using high-throughput sequencing approaches[J]. Journal of Zhejiang University-Science B, 2017, 18(4): 289-302. DOI:10.1631/jzus.b1600148.
[16] 邓风, 王玉荣, 尚雪娇, 等. 恩施地区腊肠的细菌多样性[J]. 肉类研究, 2018, 32(9): 18-22. DOI:10.7506/rlyj1001-8123-201809004.
[17] 王玉荣, 沈馨, 董蕴, 等. 鲊广椒细菌多样性评价及其对风味的影响[J]. 食品与机械, 2018, 34(4): 25-30. DOI:10.13652/j.issn.1003-5788.2018.04.005.
[18] 郭壮, 葛东颖, 尚雪娇, 等. 退化和正常窖泥微生物多样性的比较分析[J]. 食品工业科技, 2018, 39(22): 93-98. DOI:0.13386/j.issn1002-0306.2018.22.018.
[19] YANG Chengcong, ZHAO Feiyan, HOU Qiangchuan, et al. PacBio sequencing reveals bacterial community diversity in cheeses collected from different regions[J]. Journal of Dairy Science, 2020, 103(2): 1238-1249. DOI:10.3168/jds.2019-17496.
[20] CAPORASO J G, BITTINGER K, BUSHMAN F D, et al. PyNAST: a flexible tool for aligning sequences to a template alignment[J]. Bioinformatics, 2010, 26(2): 266-267. DOI:10.1093/bioinformatics/btp636.
[21] EDGAR R C. UPARSE: highly accurate OTU sequences from microbial amplicon reads[J]. Nature Methods, 2013, 10: 996. DOI:10.1038/nmeth.2604.
[22] EDGAR R C, HAAS B J, CLEMENTE J C, et al. UCHIME improves sensitivity and speed of chimera detection[J]. Bioinformatics, 2011, 27(16): 2194-2200. DOI:10.1093/bioinformatics/btr381.
[23] DESANTIS T Z, HUGENHOLTZ P, LARSEN N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB[J]. Applied and Environmental Microbiology, 2006, 72(7): 5069-5072. DOI:10.1128/aem.03006-05.
[24] MAIDAK B L, COLE J R, LILBURN T G, et al. The RDP (ribosomal database project) continues[J]. Nucleic Acids Research, 2000, 28(1): 173-174. DOI:10.1093/nar/28.1.173.
[25] MONIKA B, HUSON D H. SILVA, RDP, Greengenes, NCBI and OTT- how do these taxonomies compare?[J]. BMC Genomics, 2017, 18(Suppl 2): 114. DOI:10.1186/s12864-017-3501-4.
[26] PRICE MORGAN N, DEHAL PARAMVIR S, ARKIN ADAM P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix[J]. Molecular Biology and Evolution, 2009, 26(7): 1641-1650. DOI:10.1093/molbev/msp077.
[27] WILKINSON T J, HUWS S A, EDWARDS J E, et al. CowPI: a rumen microbiome focussed version of the PICRUSt functional inference software[J]. Frontiers in Microbiology, 2018, 9(5): 1095. DOI:10.3389/fmicb.2018.01095.
[28] 姚国强. 传统发酵乳中细菌多样性及其功能基因研究[D]. 呼和浩特: 内蒙古农业大学, 2017.
[29] XIE Mengxi, AN Feiyu, ZHAO Yue, et al. Metagenomic analysis of bacterial community structure and functions during the fermentation of da-jiang, a Chinese traditional fermented food[J]. LWT-Food Science and Technology, 2020, 30(4): 109450. DOI:10.1016/j.lwt.2020.109450.
[30] STADLMAYR A, FENG Qiang, HUBER-SCH?NAUER U, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence[J]. Nature Communications, 2015, 6: 6528. DOI:10.1038/ncomms7528.
[31] 張筠, 孟祥晨. 乳酸菌的胁迫应答及其对碳水化合物代谢的影响[J]. 中国食品学报, 2017, 17(6): 145-151. DOI:10.16429/j.1009-7848.2017.06.020
