磁性金属有机骨架-聚多巴胺的制备并用于水产养殖水样中阳离子染料的萃取检测
2020-12-25杨晓涵吴雯倩吴小海
杨晓涵,吴雯倩,王 彬,林 风,吴小海,卢 昕
(药用资源化学与药物分子工程教育部重点实验室,广西师范大学化学与药学学院,广西 桂林 541004)
染料是水生生态系统中的众多污染物之一,一些染料及其降解产物具有很高的动物细胞毒性。水溶性的阳离子碱性染料,如三苯甲烷类的亚甲蓝(methylene blue,MB)、结晶紫(crystal violet,CV)等,不仅可用于纺织业,还常被用作水产养殖业的杀菌剂[1,2]。CV可通过食入和皮肤吸收进入鱼类体内,随即转化为隐色CV残留在生物体内且难于排出。目前确知CV具有致癌高风险,还能刺激皮肤和消化道,甚至可能导致呼吸和肾功能衰竭及永久性失明[3,4]。美国、欧盟、加拿大、中国等国家和地区均未批准在水产养殖中使用CV[5,6]。MB的毒性虽不及CV,但仍可使人出现心率加快、呕吐、休克、紫绀、黄疸等症状[7]。因此,对环境水体及淡水养殖用水中痕量染料的检测监控意义重大,而富集除杂是整个分析程序中必须进行的第一步。

酚类化合物因其具有金属螯合、氢键、静电作用、pH响应等性质,使其在制备功能化材料方面成为一类独特的结构单元[28-30]。许多金属离子(如Fe、Co、Zn等)可以通过螯合作用与天然多酚化合物形成金属-酚醛网络结构[31]。利用这一性质,可将一些多酚类物质作为MOFs的蚀刻剂并使其表面功能化。Hu等[32]设计了一种利用酚酸(没食子酸或单宁酸)释放的质子破坏MOFs(如ZIF-8、ZIF-67等)的框架结构,使MOFs内部产生更大空隙,同时对其进行表面修饰的策略,并解释了由于多酚酸分子具有较大的尺寸仅能附着于MOFs的表面,一方面可以防止MOFs被完全蚀刻,同时还能使其表面的疏水性转变为亲水性,获得具有固定晶体骨架的空心MOFs晶体。Brinker等[33]通过调节pH条件和蚀刻时间来制备具有可控形态、尺寸和粗糙度的中空纳米结构,并提出了一种基于酚类脂质分子通过相转移过程快速使MOFs外层表面功能化的新策略[34]。
多巴胺(dopamine,DA)分子中同时含有儿茶酚基团和氨基,因此,既具有上述酚酸对MOFs的蚀刻功能,又可以在温和的条件下自聚合形成聚多巴胺(polydopamine,PDA)涂层[35-37]。同时,PDA中丰富的儿茶酚基团可以与MOFs晶体中的金属离子置换螯合,形成金属-聚多巴胺孔洞结构[28,38]。因此,PDA及其衍生出的复合材料已经作为吸附剂成功地用于样品前处理[39]。Huang等[40]将PDA及其衍生的复合材料作为MSPE萃取剂用于富集雌性激素;McCullum等[41]将PDA包裹的Fe3O4核壳磁性纳米粒子用于富集液体食品中的黄曲霉毒素;Li课题组[42]和Wang等[43]也利用PDA成功制备磁性纳米粒子用于富集、检测环境水样中的4种酚类化合物和多环芳烃。
本工作以预先封装了Fe3O4纳米粒子的ZIF-67为模板,利用DA分子中的儿茶酚基团螯合ZIF-67晶体中的Co2+进行蚀刻扩孔,并发生自聚合,形成具有孔洞结构的PDA亲水性外壳,制备了磁性Fe3O4-MOFs-PDA(Fe3O4@Z67D)新型材料。以碱性染料MB和CV作为目标分子,验证了Fe3O4@Z67D作为MSPE吸附剂的适用性,结合高效液相色谱技术,建立了一种简单灵敏的分析方法,成功用于检测淡水鱼养殖用水中痕量的MB和CV。
1 实验部分
1.1 实验材料和试剂
乙腈(HPLC/ACS)、甲醇(HPLC/ACS)和聚乙二醇(PEG)6000均购买于北京百灵威公司。三羟甲基氨基甲烷(>99.9%)来自英国Apollo科技有限公司。MB购买于上海萨恩化学技术有限公司。CV来自于上海阿拉丁生化科技股份有限公司。2-甲基咪唑购自上海麦克林生化有限公司。六水合硝酸钴及其他基础试剂均购自广州西陇科学股份有限公司。实验用水为Merck Millipore(美国Bedford公司)中获得的超纯水(18.2 MΩ·cm)。
标准储备溶液MB和CV(1 g/L)用甲醇配制,操作液在使用前以水稀释至所需浓度。

图1 Fe3O4@Z67D的(a)合成示意图及(b)磁固相萃取流程图Fig.1 Schemes of (a)synthesis of Fe3O4@Z67D and (b)MSPE processPEG:polyethylene glycol;EG:ethylene glycol;PDA:polydopamine;MSPE:magnetic solid-phase extraction.
1.2 表征和分析仪器
X射线衍射测定使用D/Max-3c仪器(日本Rigaku公司)。红外光谱测定使用铂金埃尔默Spectrum Two(美国PE公司)。所合成材料的形貌及元素组成通过QUANTA250扫描电子显微镜、Tecnai G2 F20 S-TWIN透射电子显微镜观测(美国FEI公司)。X射线光电子能谱分析在ESCALAB 250XI仪器(美国Thermo Fisher公司)上进行。Brunauer-Emmett-Teller表面积由Autosorb-IQ仪器(美国Quantachrome公司)在77 K下收集的N2吸附-解吸等温线计算得到。超导量子干涉磁力计(MPMS,7T-SQUID XL,美国QuantumDesign公司)在300 K下记录样品的磁性。日本Shimadzu公司的10ATVP高效液相色谱仪进行色谱分析,配套设备包括SPD-10AVP紫外检测器、LC-10AT二元高压梯度泵、CLASS-VP 5.03色谱工作站。
1.3 Fe3O4@Z67D的合成
Fe3O4@Z67D的制备过程如图1a所示。
Fe3O4的合成:分别称取0.68 g FeCl3、0.1 g柠檬酸钠、1.8 g乙酸钠、0.5 g PEG 6000于烧杯中,加入20 mL乙二醇溶解,转移至200 ℃反应釜中反应10 h。冷却至室温,产物用甲醇和水交替洗涤多次,干燥后得到Fe3O4。
Fe3O4@ZIF-67的合成:称取0.2 g上述的Fe3O4,以及Co(NO3)2·6H2O(5.82 g,20 mmol)和2-甲基咪唑(6.57 g,80 mmol),分别溶于50 mL甲醇。将Co(NO3)2溶液加入Fe3O4分散液中,搅拌60 min。加入2-甲基咪唑溶液,室温下搅拌24 h后,收集紫色的Fe3O4@ZIF-67并用甲醇洗涤数次,干燥待用。
Fe3O4@Z67D的合成:称取0.2 g干燥的Fe3O4@ZIF-67,分散在20 mL乙醇和30 mL水中,加入0.01 g盐酸多巴胺,搅拌5 min。再加入150 mL Tris-HCl (pH 7.2),室温下搅拌12 h。收集棕色固体,用水和甲醇洗涤多次,60 ℃下干燥。
1.4 磁固相萃取流程
磁固相萃取过程见图1b。将15 mg Fe3O4@Z67D加入200 mL样品溶液中,超声10 min后,磁铁分离。将吸附剂全部转移至小烧杯中,加入8.0 mL 0.1%(v/v)乙酸甲醇(4.0 mL/次),超声15 min洗脱目标物。磁铁分离后收集上清液并用氮气吹干。残留物重溶于150 μL流动相中,0.22 μm针式滤器过滤,取20.0 μL注入HPLC进行分析。
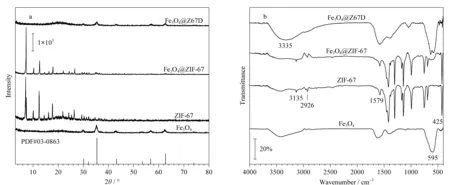
图2 Fe3O4、ZIF-67、Fe3O4@ZIF-67和Fe3O4@Z67D的(a)X射线衍射谱图和(b)红外光谱图Fig.2 (a)X-ray diffraction spectra and (b)infrared spectra of Fe3O4,ZIF-67,Fe3O4@ZIF-67 and Fe3O4@Z67D
1.5 实际样品的采集处理
实际水样采集于广西桂林市郊区的养殖鱼塘,0.45 μm滤膜过滤后于4 ℃冰箱保存待用。
1.6 HPLC分析
样品分离采用SpursilTMC18色谱柱(250 mm×4.6 mm,5 μm,中国Dikma公司)。流动相由甲醇(A)和0.012 5 mol/L乙酸铵缓冲溶液(乙酸调pH至4.5)(B)组成,A与B的体积比为85∶15,流速为1.0 mL/min,柱温为35 ℃,检测波长分别为290 nm(MB)、580 nm(CV)。
2 结果与讨论
2.1 Fe3O4@Z67D的表征
2.1.1X射线衍射分析

2.1.2红外光谱分析
红外光谱能进一步说明Fe3O4@Z67D被蚀刻,释放2-甲基咪唑,形成Fe3O4@Z67D新型材料。如图2b所示,在Fe3O4、Fe3O4@ZIF-67和Fe3O4@Z67D中均含有Fe-O伸缩振动峰(600 cm-1),说明Fe3O4成功合成[48]。ZIF-67中的金属Co离子是与2-甲基咪唑中的N配位(425 cm-1,Co-N),而经蚀刻后,Co-N吸收峰几乎消失,说明金属Co离子由原来与N配位转移与多巴胺中的O配位,即ZIF-67的结构大部分被破坏[30,45];此外,Fe3O4@Z67D的谱图中,在3 330 cm-1附近出现N-H和O-H伸缩振动的吸收峰,这也证明了在蚀刻的过程中,DA氧化聚合形成PDA,此结果与X射线衍射结果一致。
2.1.3电镜分析
为观察所合成材料的形貌并进一步分析其元素组成,进行了扫描电镜、透射电镜及能量色散X射线光谱分析,结果见图3和4。

图3 (a)Fe3O4、(b)Fe3O4@ZIF-67和(c)Fe3O4@Z67D的扫描电镜图,以及(d)Fe3O4@Z67D和(e,f)Fe3O4@Z67D的透射电镜图Fig.3 Scanning electron microscope images of (a)Fe3O4,(b)Fe3O4@ZIF-67,(c)Fe3O4@Z67D and transmission electron microscopy images of (d)Fe3O4@ZIF-67 and (e,f)Fe3O4@Z67D
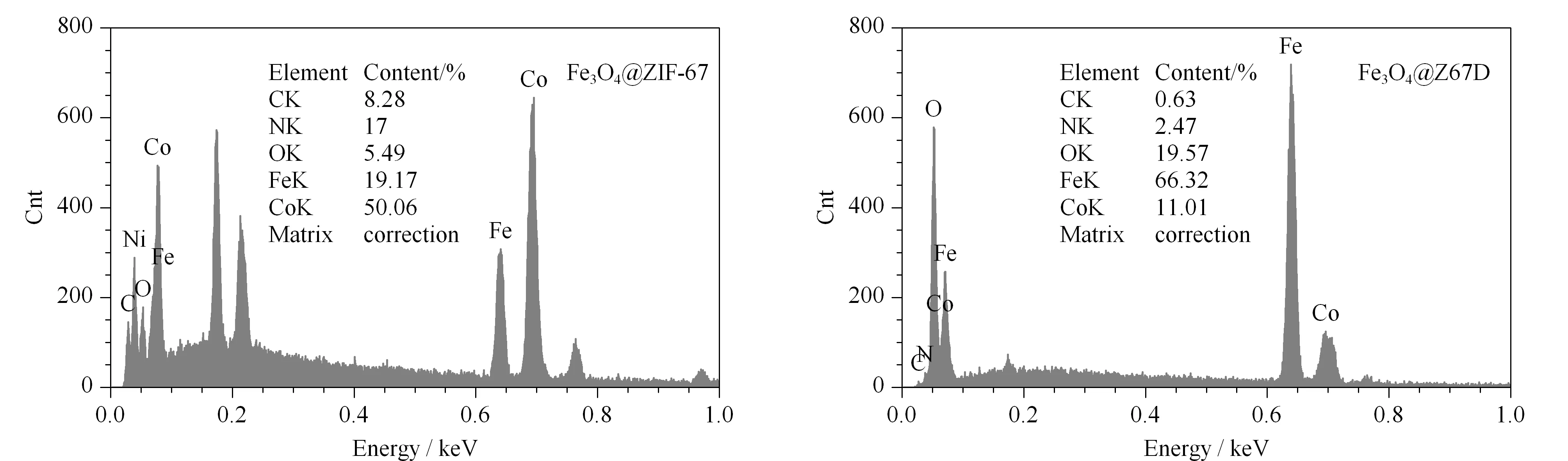
图4 (a)Fe3O4@ZIF-67和(b)Fe3O4@Z67D的能量色散X射线光谱图Fig.4 Energy dispersive X-ray spectra of (a)Fe3O4@ZIF-67 and (b)Fe3O4@Z67D
用水热法制备的Fe3O4,其形状为球形,粒径为10~50 nm(见图3a)。Fe3O4@ZIF-67具备ZIF-67的菱形十二面体形状,表面较光滑(见图3b),并在图3d中清晰可见其内部封装的Fe3O4纳米粒子。在加入DA后,Fe3O4@ZIF-67的菱形消失,形成球形的Fe3O4@Z67D(见图3c和3e),且粒径为100~200 nm,证明DA蚀刻破坏了大部分ZIF-67的结构而非直接包裹。颗粒外层浅色结构部分为DA氧化聚合后形成的PAD(见图3f),这与X射线衍射及红外光谱的结果一致。图4的能谱分析也进一步证实了蚀刻的发生,即Fe3O4@ZIF-67和Fe3O4@Z67D中虽含有相同的元素,但后者的Co元素含量降低,O含量增加,这也是因为PDA破坏了大部分ZIF-67的配位键,释放出2-甲基咪唑配体及Co离子所致,而在Fe3O4@Z67D中含有少量的N元素,还存在少量的ZIF-67,说明DA主要蚀刻ZIF-67,而非腐蚀。
2.1.4X射线光电子能谱分析
为进一步验证Fe3O4@Z67D的化学组成,实验测量了X射线光电子能谱,结果见图5。

图5 Fe3O4@Z67D的(a)X射线光电子能谱全扫描分析及(b)Fe 2p、(c)Co 2p、(d)O 1s、(e)C 1s和(f)N 1s的高分辨X射线光电子能谱分析Fig.5 X-ray photoelectron spectroscopy full-scan analysis of (a)Fe3O4@Z67D and high resolution X-ray photoelectron spectroscopy analysis of (b)Fe 2p,(c)Co 2p,(d)O 1s,(e)C 1s and (f)N 1s
图5a中的全扫描能谱中显示,Fe3O4@Z67D中存在C、O、N、Fe和Co元素,与图4一致。对各元素的高分辨能谱图进行分峰与归属,可得以下结论。Fe 2p能谱图(见图5b)中,峰值为711.27 eV和724.07 eV的峰可分别归属于Fe 2p3/2和Fe 2p1/2,由于二价Fe(2p3/2)和三价Fe(2p1/2)的出现,证明了Fe3O4@Z67D中存在Fe3O4[48,49]。在图5c的Co 2p能谱图中,781.09 eV和796.89 eV处的两个主峰分别归属于Co 2p3/2和Co 2p1/2,两个卫星峰分别位于785.13 eV和802.40 eV处。由于主峰与卫星峰的能隙在4~5.5 eV,是二价Co离子的能隙,证实Fe3O4@Z67D中的Co元素主要以二价Co形式存在[50]。O 1s能谱图(见图5d)中位于529.95、531.34和532.65 eV处的3个峰分别对应于Fe3O4@Z67D中金属氧化物Fe-O、聚多巴胺中的-OH和C=O键。图5e的C 1s能谱可拟合为C-C/C=C(284.62 eV)、C-N/C=N(286.03 eV)和C=O(288.25 eV)3个峰。从图5f的N 1s能谱图中,可观察到在399.86 eV和398.53 eV处的峰,对应2-甲基咪唑和聚多巴胺中的-NH键以及聚多巴胺中的C=N键[51,52]。以上结果与图2b一致,因此进一步证明了Fe3O4@Z67D材料的成功合成。
2.1.5氮吸附-解吸分析
为考察材料的比表面积,对Fe3O4@ZIF-67和Fe3O4@Z67D进行了氮气吸附-解吸分析,结果如图6所示。经对比分析可知,Fe3O4@ZIF-67的比表面积为1 337 m2/g,平均孔径在4 nm左右。经多巴胺蚀刻后,Fe3O4@Z67D的比表面积下降至93 m2/g,孔径增大到110 nm,还依然保留少量4 nm孔径的ZIF-67。此结果和图4一致,进一步证实了ZIF-67起到“牺牲”模板和桥梁的作用,导致蚀刻、扩孔过程的发生,从而形成多孔Z67D。这将有利于目标分子在新型材料孔洞内的扩散。
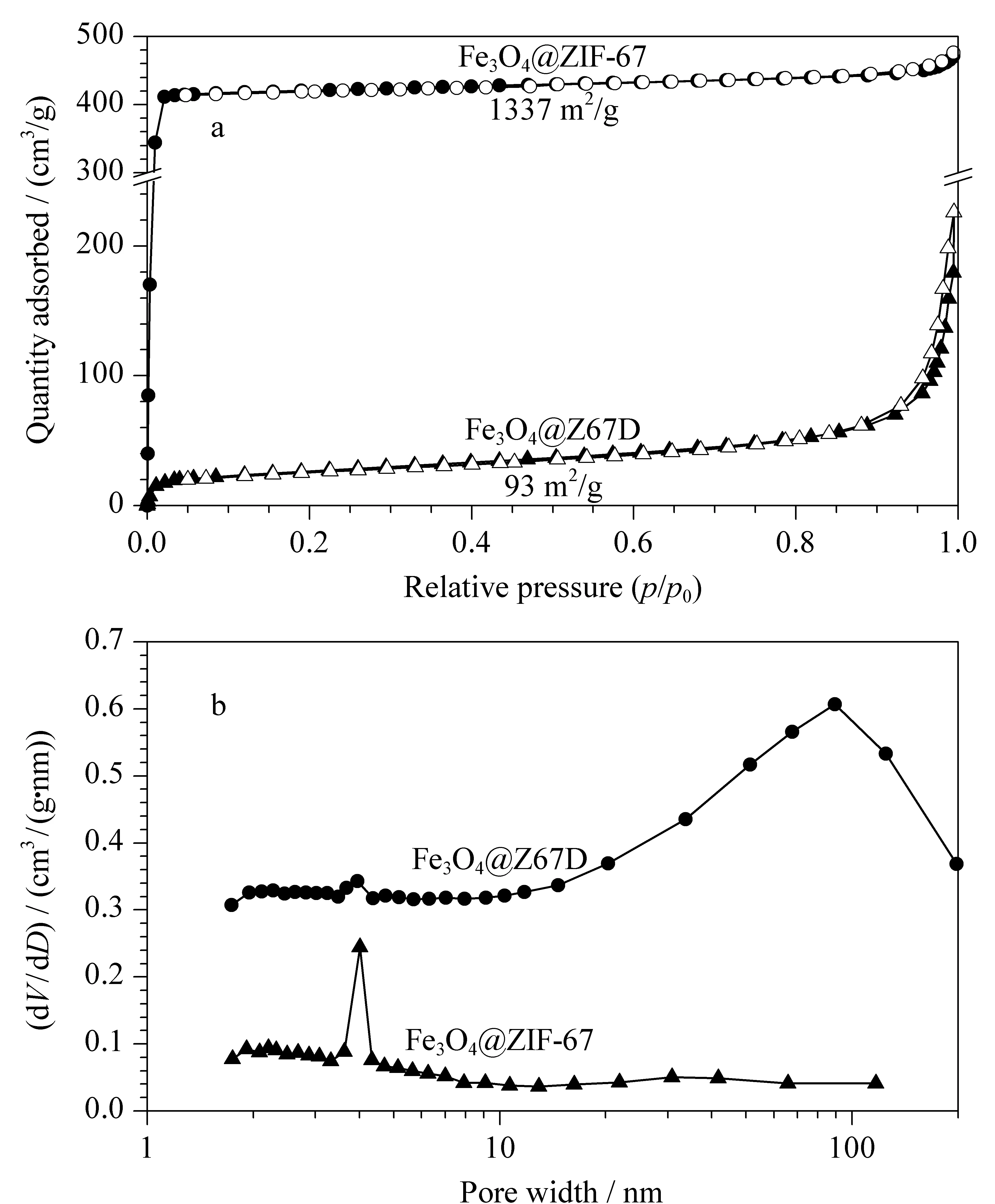
图6 Fe3O4@ZIF-67和Fe3O4@Z67D的(a)N2吸附-解吸等温线及(b)孔径分布Fig.6 (a)N2 adsorption-desorption isotherm and (b)pore size distribution of Fe3O4@ZIF-67 and Fe3O4@Z67D
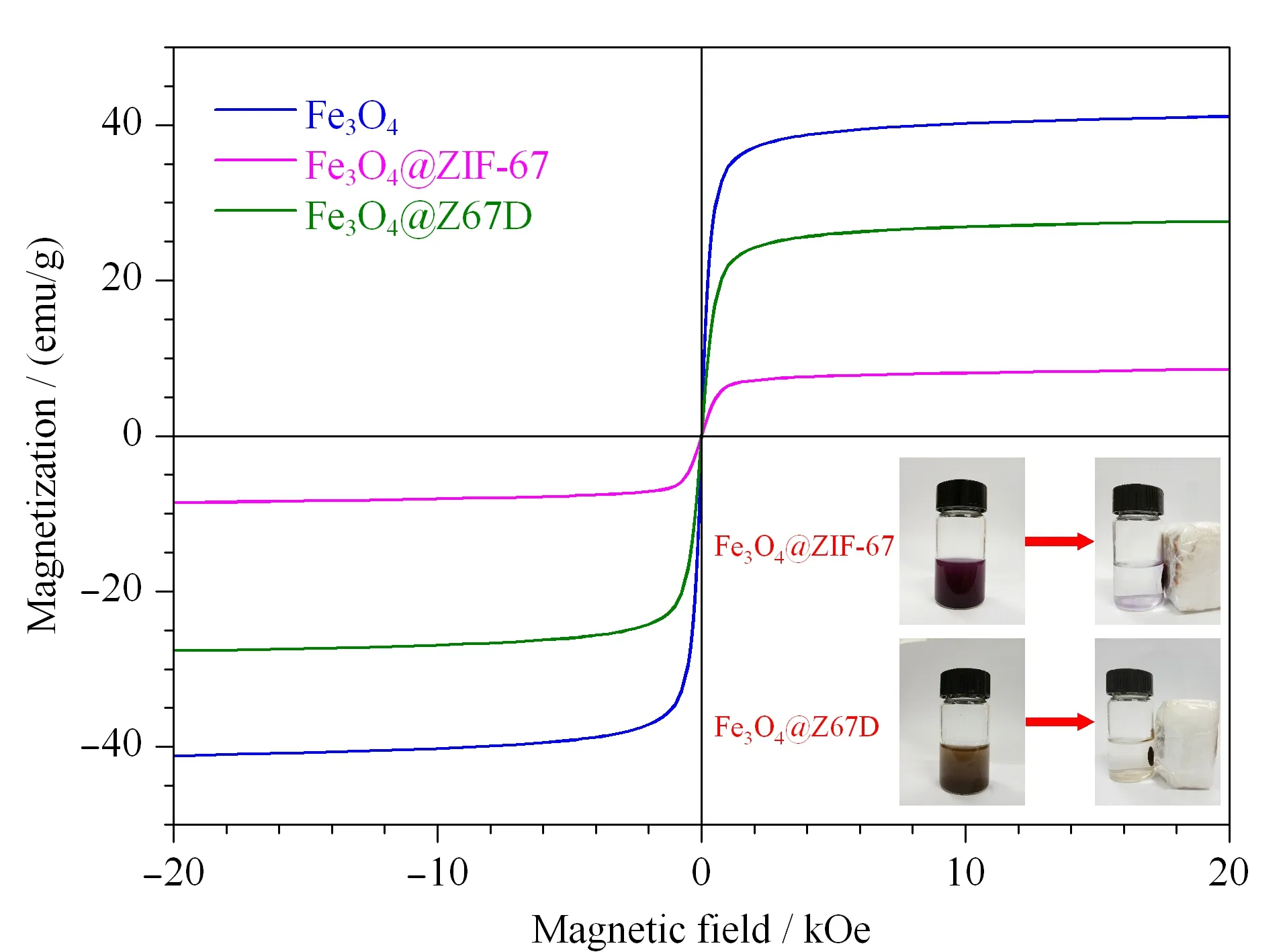
图7 Fe3O4,Fe3O4@ZIF-67 and Fe3O4@Z67D的磁滞回线Fig.7 Hysteresis loops of Fe3O4,Fe3O4@ZIF-67 and Fe3O4@Z67D
2.1.6磁性测量
新型材料是否适用于磁分离,需进行磁滞回线测定,结果见图7。图中显示了Fe3O4的强磁性在被封装于ZIF-67晶体中后,磁化强度由41.1 emu/g大幅度降低至8.6 emu/g。但Fe3O4@Z67D形成后,蚀刻和聚合的发生使磁化强度又恢复至27.6 emu/g。这已完全能够满足磁分离的需要。
综上所述,多种表征手段充分证实,DA蚀刻大部分的ZIF-67,导致其特有晶型被破坏,并能与非游离的金属Co2+螯合,氧化聚合成PDA,形成Fe3O4@Z67D新型材料,N2吸附-解吸性能优异,磁性保持良好。这都将为其应用于MSPE提供保障。
2.2 萃取条件的优化
以MB和CV为目标分子,评估了Fe3O4@Z67D材料的MSPE适用性。初步证实其对两种染料具有良好的吸附与脱附能力。其通过π-π作用和亲水作用吸附目标分析物。随后,进一步优化了影响萃取性能的多个实验参数,包括材料的用量、萃取时间、样品溶液体积、溶液pH和解吸条件等。考虑到新方法将应用于复杂水样测定,多样性的共存物质可能将与目标物发生吸附竞争,因此,优化实验将两种染料的质量浓度均控制在200 μg/L的高水平上,旨在尽可能模拟实际样品。

图8 (a)Fe3O4@Z67D吸附剂的用量、(b)萃取时间,以及样品溶液的(c)体积、(d)pH和洗脱剂(e)类型、(f)体积对两种染料回收率的影响(n=3)Fig.8 Effects of (a)the amount of Fe3O4@Z67D,(b)the extraction time,(c)the volume and (d)pH of sample solution,and (e)the type and (e)volume of the eluent solvents on the recoveries of the two dyes (n=3)MeOH:methanol;ACN:acetonitrile;HAc:acetic acid.
在200 mL合成样品溶液中考察了不同剂量(5~30 mg)的Fe3O4@Z67D对染料萃取效率的影响,结果见图8a。当Fe3O4@Z67D的用量在15 mg时,染料的回收率达到最大,之后略有下降。其原因是,吸附剂用量过多时,易发生自身团聚而降低吸附效率。或用量过多时,洗脱剂的用量相对不足,难以保证目标物完全脱附,从而导致回收率下降。因此,随后的实验中选择Fe3O4@Z67D用量为15 mg。
为了获得染料的最佳萃取时间,将Fe3O4@Z67D分散在200 mL样品溶液中,平衡5~60 min。图8b显示,MB在15 min后的回收率基本保持不变,但CV的则需要45 min才达到最高值。因此,综合考虑后选择最佳的萃取时间为45 min。
在最优的萃取剂用量下,测定了样品溶液体积在100~500 mL范围内的回收率,结果如图8c所示。当MB的溶液体积在100~300 mL时,其回收率良好并且几乎保持不变。而CV在溶液体积为300 mL时,回收率有所下降。因此,综合考虑后选择样品溶液体积为200 mL作为最佳的样品溶液体积。
样品溶液的pH环境是影响萃取效率的重要参数。用缓冲液将样品溶液的pH控制在4~8的范围内,测定染料的回收率。结果显示两种染料对萃取富集酸度环境要求有所不同(见图8d)。原因为:Fe3O4@Z67D与MB之间的作用以静电力为主,因此,易受酸性条件的影响。随着pH的升高,与Fe3O4@Z67D的静电作用增强,即材料对MB的吸附增强。而CV具有共轭大π键结构,与Fe3O4@Z67D之间的π-π共轭作用大于静电作用,因此,pH对CV的吸附效果影响不大。经过折中考虑,在后继实验中控制样品溶液的pH为7。
解吸步骤是分析物从吸附剂中释放的过程。选择适当的解吸溶剂、解吸液体积及解吸时间对分析物的解吸效率至关重要。实验考察了多种解吸溶剂的解吸效率,包括甲醇、乙腈、甲醇/乙腈(1∶1,v/v)、甲醇/乙腈(3∶1,v/v)和0.1%(v/v)乙酸甲醇。由图8e可以明显地看出,当使用0.1%(v/v)乙酸甲醇作为洗脱剂时,两种染料的回收率均达到90%以上,洗脱效果最好。继续考察上述洗脱剂的使用体积(4~12 mL)对回收率的影响,结果见图8f。综合考虑,选择8 mL 0.1%(v/v)乙酸甲醇作为最佳体积。另外,解吸时间由实验确定为15 min。
2.3 Fe3O4@Z67D的再生稳定性
吸附剂的再生稳定性是评估吸附材料性能的关键因素之一,涉及应用成本问题。本实验采用15 mg Fe3O4@Z67D萃取染料,整个吸附-解吸过程循环进行10次。从图9中可以明显看出,Fe3O4@Z67D材料重复使用10次后,染料的回收率没有出现明显变化。由此证实,Fe3O4@Z67D具有优异的再生能力和重复使用性。

图9 Fe3O4@Z67D重复使用后的回收率(n=3)Fig.9 Recoveries obtained on Fe3O4@Z67D after reuse(n=3)
2.4 方法性能的评估
在最佳的MSPE条件和色谱分离条件下,对两种染料的测定方法进行了评估。从表1可以看出,萃取后两种染料的线性范围分别为0.5~200 μg/L、0.01~50 μg/L,检出限(LOD,S/N=3)分别为0.04 μg/L、0.008 μg/L,测定的相对标准偏差(RSD,n=5)分别为1.2%、3.6%,富集因子分别为777、688。上述结果证实了Fe3O4@Z67D对MB和CV有良好的萃取效率,可以显著提高HPLC方法的灵敏度。

表1 MSPE-HPLC测定两种染料的方法学参数Table 1 Analytical performance data of the MSPE-HPLC method for the determination of the two dyes
2.5 实际样品的测定
将上述MSPE-HPLC方法进一步应用于养殖鱼塘水样中两种染料的测定,并考察回收率。测定结果见表2,水样及加标后水样的色谱图见图10。测定结果显示,在水样中可以检测到0.7 μg/L MB和0.2 μg/L CV。向水样中分别添加水平为1、10 μg/L MB和5、50 μg/L CV,测得加标回收率在82.0%~109.0%区间,RSD(n=5)低于2.9%。将本方法与其他测定MB和CV的方法进行比较(见表3),线性范围、回收率、LOD和RSD均令人满意。
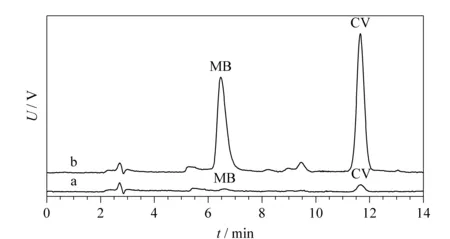
图10 MSPE处理后的鱼塘水样的色谱图Fig.10 Chromatograms obtained from fishpond water samples treated after MSPE a.blank sample;b.sample spiked with 10 μg/L MB and 5 μg/L CV. Conditions:SpursilTM C18 column(250 mm×4.6 mm,5 μm);mobile phase,methanol-0.0125 mol/L ammonium acetate buffer solution (85∶15,v/v)(pH adjusted to 4.5 by acetic acid);flow rate,1.0 mL/min;detection wavelength,580 nm;injection volume,20 μL;column temperature,35 ℃.
Y:peak area;X:mass concentrition,μg/L.
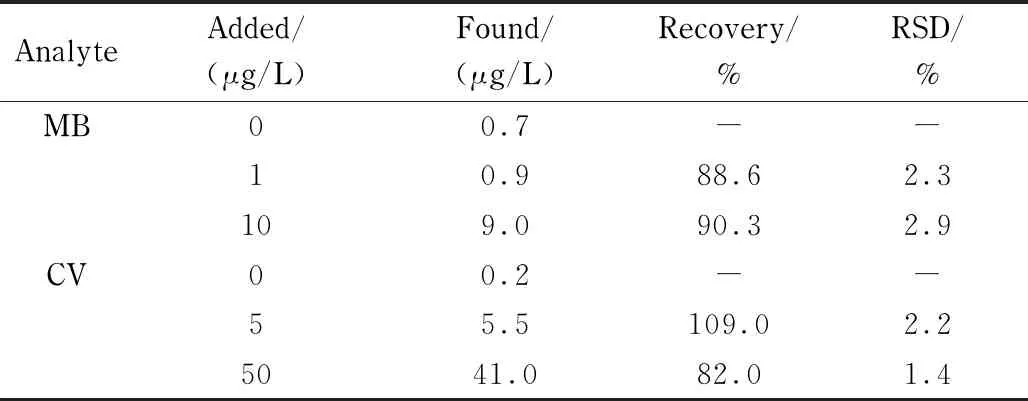
表2 养殖鱼塘水样中MB和CV的测定结果及加标回收率Table 2 Analytical results of MB and CV in fishpond water samples and the spiked recoveries (n=5)
-:not applicable.
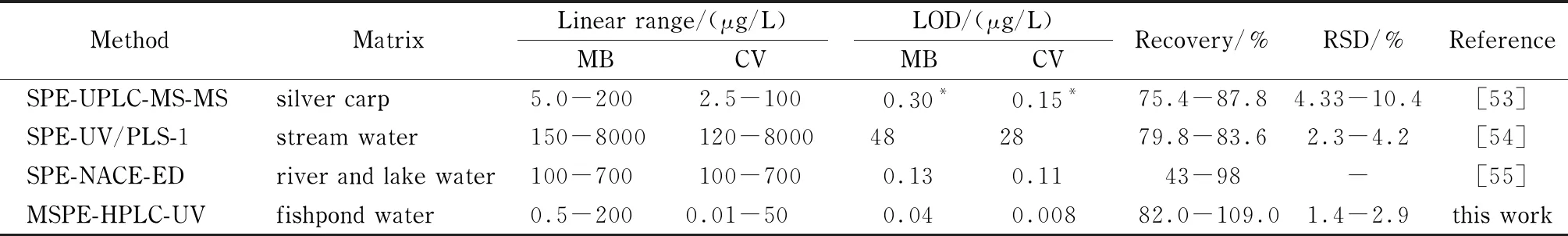
表3 本方法与相关文献中的方法比较Table 3 Comparison of this method with reported methods
PLS-1:partial least squares method;NACE:non-aqueous capillary electrophoresis;ED:electrochemical detection;-:not available;*:μg/kg.
3 结论
本实验以ZIF-67为模板,利用DA能与金属离子螯合并能够自聚合的原理,对ZIF-67进行功能化修饰,合成了新型的Fe3O4@Z67D材料。多种表征手段证实了DA能将ZIF-67结构中过小的孔径蚀刻扩大,再经自聚合后形成多孔PDA壳层,且新型材料具有良好的亲水性。以两种阳离子染料为目标分子,对Fe3O4@Z67D作为MSPE萃取剂的适用性进行考察,结果显示良好的萃取效率。材料结构稳定性尤为出色,在MSPE过程中可多次重复使用,而萃取效率不降低,这将有效降低材料使用成本。该新型材料成功应用于水产养殖水样中阳离子染料的富集萃取,成为一种高效、快速的样品前处理方法,使经典的高效液相色谱-紫外检测方法可用于某些痕量物质的检测。
