UPLC-ESI-MS/MS法同时测定强力定眩胶囊中的7个指标成分
2020-12-23边敏琦邹俊波崔春利张小飞史亚军郭东艳乔安平贾小刚
王 媚,边敏琦,邹俊波,崔春利,张小飞,史亚军,郭东艳,乔安平,贾小刚
(陕西省中药基础与新药研究重点实验室 陕西中医药大学药学院,陕西 咸阳 712046)
强力定眩胶囊(Qiangli Dingxuan Capsule,QLDXC)由天麻、杜仲、杜仲叶、野菊花和川芎等5种中药材经过一系列的工艺制成,主要用于治疗高血压、动脉硬化、高血脂等症及因其引起的头痛、头晕、目眩等。其降压作用缓和持久,降脂作用明显,特别是在眩晕患者的临床治疗中应用广泛,效果显著[1]。强力定眩胶囊是2012年国家药品标准提高品种,之前的质量标准为国家食品药品监督管理局标准YBZ03582009,今年被收录于《中华人民共和国药典》(一部)2020年版,其检验项目只有常规检验和以天麻素为指标的量化控制。文献[2-3]报道,天麻含有天麻素、对羟基苯甲醇等有效成分;杜仲含有松脂醇单葡萄糖苷、松脂醇二葡萄糖苷等有效成分;杜仲叶含有绿原酸、阿魏酸等有效成分;野菊花含有蒙花苷、阿魏酸、绿原酸等有效成分。若仅以有效成分天麻素为指标难以全面准确控制强力定眩胶囊质量,为了更好地发挥药品的临床疗效,有必要制定较为全面的强力定眩胶囊的多指标评价标准。
超高效液相色谱-电喷雾-三重四级杆质谱联用(UPLC-ESI-MS/MS)是目前广泛应用的分析方法之一[4-8],具有色谱的分离能力和质谱的高分辨性,适合分析复杂的中药多组分体系。作者采用UPLC-ESI-MS/MS方法,同时测定强力定眩胶囊中天麻素、绿原酸、阿魏酸、蒙花苷、对羟基苯甲醇、松脂醇单葡萄糖苷、松脂醇二葡萄糖苷等7个指标成分,以期为全面、有效地控制强力定眩胶囊的质量提供依据。
1 实验
1.1 材料、试剂与仪器
强力定眩胶囊,陕西汉王药业有限公司;中药材天麻、杜仲、杜仲叶、野菊花、川芎等购于西安兴盛德中药饮品有限责任公司,经鉴定均符合《中华人民共和国药典》(一部)2015年版规定。
天麻素对照品(批号AYJJ-V5AG)、绿原酸对照品(批号R1F7-9B91)、阿魏酸对照品(批号PQEG-8GF8)、蒙花苷对照品(批号TUQ4-Y52R)、对羟基苯甲醇对照品(批号DJ9T-VXN8),中国药品生物制品监督检验所;松脂醇单葡萄糖苷对照品(批号B21724)、松脂醇二葡萄糖苷对照品(批号3GCF-TDYY),上海源叶生物技术公司;甲醇、甲酸,质谱纯,德国默克公司;无水乳糖;无水乙醇(分析纯);实验用水为屈臣氏蒸馏水。
ACQUITY I-Class UPLC system-Xevo TQ-XS MS型液质联用仪,美国Waters公司;AR1140型电子分析天平,梅特勒-托利多仪器上海有限公司;DZF-6050型真空干燥箱,上海一恒科学仪器有限公司;振动式细胞级超微粉碎机,济南达微机械有限公司;RE-3000型旋转蒸发器,上海亚荣生化仪器厂;电热恒温水浴锅,北京科伟永兴仪器有限公司;QE-300型高速万能粉碎机,浙江屹立工贸有限公司;智能溶出仪。
1.2 7个指标成分的化学结构式(图1)1.3 UPLC-ESI-MS/MS条件
UPLC-ESI-MS/MS系统由ACQUITY I-Class UPLC和Xevo TQ-XS MS组成。
色谱柱为Thermo Syncronis C18柱(100 mm×2.1 mm,1.7 μm),流动相为0.1%甲酸水溶液(A)-甲醇(B),梯度洗脱程序为:0~1 min,10%B;1~5.8 min,10%B~40%B;5.8~6.3 min,40%B~95%B;6.3~8.5 min,95%B;8.5~8.7 min,95%B~10%B;8.7~10 min,10%B。柱温35 ℃,流速0.4 mL·min-1,进样量5 μL。
质谱检测器为电喷雾离子源(ESI),ESI-MS以正离子和负离子多反应监测(MRM)模式进行测试,喷雾电压3.0 kV,毛细管温度450 ℃,去溶剂气体流速1 000 μL·h-1,喷雾气体压力7.0 bar。质谱化合物离子对的去簇电压和碰撞能量采用Masslynx Ⅴ4.2 进行采集和优化。
1.4 溶液的制备
1.4.1 混合对照溶液的制备
精密称取各对照品,分别加入70%甲醇溶液配制成1.37 mg·mL-1天麻素、5.10 mg·mL-1绿原酸、1.79 mg·mL-1阿魏酸、2.64 mg·mL-1蒙花苷、5.25 mg·mL-1对羟基苯甲醇、0.98 mg·mL-1松脂醇单葡萄糖苷、0.96 mg·mL-1松脂醇二葡萄糖苷等储备液,于4 ℃保存,备用;分别吸取一定量上述各对照品储备液于5 mL容量瓶中,加入70%甲醇,定容,得到含天麻素274.000 0 μg·mL-1、绿原酸1 020.096 1 μg·mL-1、阿魏酸358.036 8 μg·mL-1、蒙花苷528.248 3 μg·mL-1、对羟基苯甲醇105.063 0 μg·mL-1、松脂醇单葡萄糖苷196.154 0 μg·mL-1、松脂醇二葡萄糖苷192.142 1 μg·mL-1的混合对照溶液,于4 ℃保存,备用。进样前,过0.22 μm微孔滤膜。
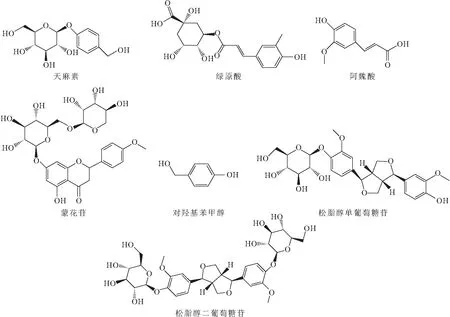
图1 7个指标成分的化学结构式Fig.1 Chemical structures of seven index compositions
1.4.2 供试溶液的制备
取适量样品研细后,精密称定适量;加入稀乙醇50 mL[2],称重;45 ℃超声处理30 min,取出冷至室温,再称重;用稀乙醇补足减失的重量,摇匀,滤液蒸干,残渣用乙腈-水(3∶97,体积比)溶解,定容至10 mL容量瓶中,摇匀,即得供试溶液。进样前,过0.22 μm微孔滤膜。
1.4.3 阴性对照溶液的制备
按照强力定眩胶囊的生产工艺,分别制备不含天麻、杜仲叶、野菊花、川芎、杜仲的空白溶液,再按1.4.2方法制备阴性对照溶液。
2 结果与讨论
2.1 混合对照溶液和供试溶液的指标成分的MRM图谱(图2)2.2 方法学考察
2.2.1 专属性
分别取混合对照溶液、供试溶液和阴性对照溶液,按色谱条件进样测定,进样量3 μL。结果显示,天麻素、绿原酸、阿魏酸、蒙花苷、对羟基苯甲醇、松脂醇单葡萄糖苷、松脂醇二葡萄糖苷等指标成分的测定不受其它干扰,峰型良好。表明处方中的其它成分对测定结果无影响。
2.2.2 线性关系
精密吸取混合对照溶液,逐级稀释5倍,加入一定量的甲醇,制备6种不同浓度梯度的混合对照溶液,按MRM模式进样测定,进样量3 μL。以各待测成分的峰面积(y)为纵坐标、浓度(x,μg·mL-1)为横坐标绘制标准曲线。
将上述不同浓度梯度的混合对照溶液加入到空白样品溶液中,按最优实验条件进样测定,以峰高除以背景噪声值作为S/N,计算检出限(LOD,S/N≥3)和定量限(LOQ,S/N≥10),结果见表1。

图2 混合对照溶液(a)和供试溶液(b)的指标成分的MRM图谱Fig.2 MRM chromatograms of index compositions from mixed reference solution(a) and test solution(b)

表1 强力定眩胶囊指标成分的线性方程、检出限和定量限
2.2.3 精密度
精密吸取混合对照溶液0.2 mL,用甲醇定容于10 mL容量瓶中,摇匀,制成含天麻素、绿原酸、阿魏酸、蒙花苷、对羟基苯甲醇、松脂醇单葡萄糖苷、松脂醇二葡萄糖苷分别为5.48 μg·mL-1、 20.40 μg·mL-1、7.16 μg·mL-1、10.56 μg·mL-1、21.00 μg·mL-1、 3.92 μg·mL-1、3.84 μg·mL-1的对照溶液,按色谱条件连续进样6次。结果显示,指标成分响应信号的 RSD 为0.89%~5.94%,保留时间的 RSD为0.07%~0.22%。表明仪器精密度良好。
2.2.4 重复性
取强力定眩胶囊内容物(批号190505)6份,按1.4.2方法制备供试溶液,按色谱条件进样测定,记录各指标成分的信号强度,计算质量分数。结果显示,指标成分响应信号的RSD为1.18%~3.11%,保留时间的RSD为0.21%~0.32%。表明该方法重复性良好。
2.2.5 稳定性
取强力定眩胶囊内容物,按1.4.2方法制备供试溶液,分别在0 h、4 h、10 h、16 h、24 h取样测定。结果显示,指标成分响应信号的RSD为0.84%~3.06%,保留时间的RSD为0.18%~0.37%。表明供试溶液在24 h内稳定。
2.2.6 加标回收率
精密称取强力定眩胶囊内容物(批号190505)6份,每份0.1 g,按1.4.2方法制备供试溶液,加入相当于已知量的100%的对照溶液,进样测定,进样量3 μL,记录各指标成分的峰面积,计算加标回收率,结果见表2。
由表2可知,强力定眩胶囊各指标成分的平均加标回收率为98.99%~101.92%,RSD为0.37%~1.54%。

表2 强力定眩胶囊指标成分的加标回收率(n=6)
2.3 样品测定
取不同批号的强力定眩胶囊内容物,按1.4.2方法制备供试溶液,按色谱条件进样测定,计算指标成分的含量,结果见表3。

表3 5批强力定眩胶囊指标成分的测定结果(mg,n=3)
2.4 讨论
2.4.1 提取条件的优化
为了使强力定眩胶囊中的天麻素、对羟基苯甲醇、松脂醇单葡萄糖苷等多种指标成分得到充分提取,结合赵斌等[3]报道的方法,以稀乙醇为提取溶剂,在不同温度下超声提取不同时间,考察超声时间对强力定眩胶囊中指标成分提取率的影响。结果显示,在超声时间为30 min时,强力定眩胶囊中的指标成分提取率最高;在超声温度为45 ℃时,强力定眩胶囊中的指标成分提取率较高。因此,选择超声时间和超声温度分别为30 min和45 ℃。
2.4.2 色谱条件的优化
分别考察了甲酸水溶液-乙腈、甲酸水溶液-甲醇流动相对强力定眩胶囊中指标成分分离效果的影响。结果表明,在0.1%甲酸水溶液-甲醇体系下进行梯度洗脱时,对羟基苯甲醇和蒙花苷等指标成分的响应信号增强,离子化程度更好,峰形美观,无其它干扰。
同时,分别考察了Thermo Syncronis C18柱(100 mm×2.1 mm, 1.7 μm)、岛津XR-ODS C18柱(75 mm×2.1 mm,2.2 μm)、Agilent 5 TC-C18柱(250 mm×4.6 mm,5 μm)、Waters BEH C18 柱(50 mm×2.1 mm,1.7 μm)、COSMOSIL 5 C18-MS-Ⅱ柱(150 mm×4.6 mm,5 μm)等色谱柱对强力定眩胶囊指标成分分离效果的影响。结果表明,Thermo Syncronis C18柱(100 mm×2.1 mm,1.7 μm)的分离效果最好,分析速度快(8 min内),大大提高了分析效率。因此,选用Thermo Syncronis C18柱(100 mm×2.1 mm,1.7 μm)。
2.4.3 质谱条件的优化
实验质谱条件采用MRM模式,根据指标化合物母离子与子离子的质荷比信息,对质谱的去簇电压、碰撞能量和碰撞池出口电压等参数逐一进行优化(表4),得到的化合物信息特异性、灵敏度和准确性都很高。
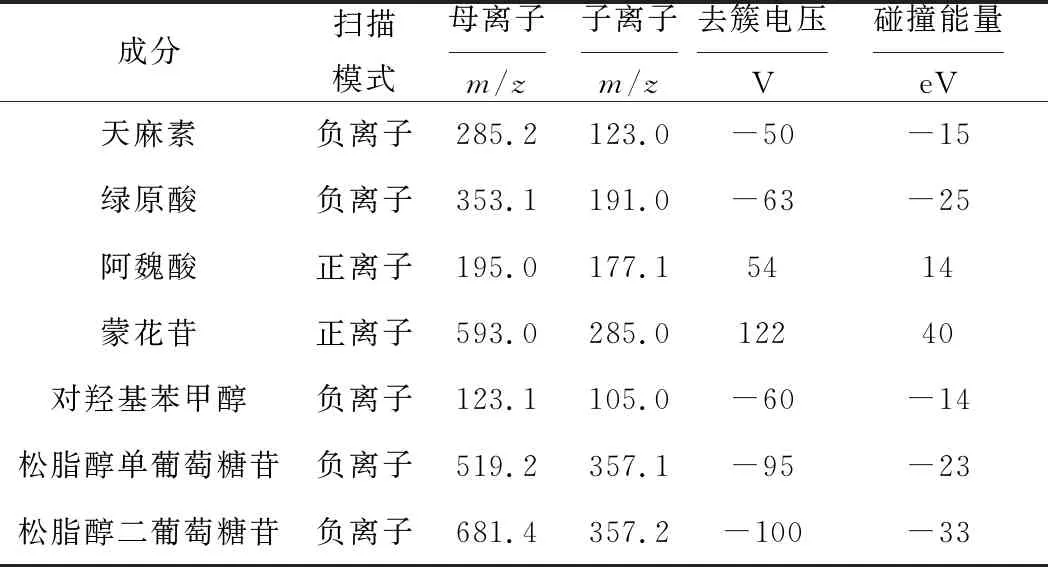
表4 强力定眩胶囊指标成分的质谱条件
3 结论
建立了同时测定强力定眩胶囊中天麻素、绿原酸、阿魏酸、蒙花苷、对羟基苯甲醇、松脂醇单葡萄糖苷、松脂醇二葡萄糖苷的UPLC-ESI-MS/MS方法。该方法操作简便,选择性好,灵敏度高,分析速度快,结果准确,为有效控制强力定眩胶囊的质量提供了保障,为多成分全面评价强力定眩胶囊质量提供了新思路。
