Role of platelets and breast cancer stem cells in metastasis
2020-12-22GretelMendozaAlmanzaLuisBurciagaHernndezVilmaMaldonadoJorgeMelendezZajglaJorgeOlmos
Gretel Mendoza-Almanza,Luis Burciaga-Hernández, Vilma Maldonado,Jorge Melendez-Zajgla, Jorge Olmos
Gretel Mendoza-Almanza,CONACYT, Universidad Autónoma de Zacatecas., Zacatecas 98068, Mexico
Luis Burciaga-Hernández,Maestría en Ciencias Biomédicas, Universidad Autónoma de Zacatecas, Zacatecas 98160, Mexico
Vilma Maldonado,Laboratorio de Epigenética, Instituto Nacional de Medicina Genómica, Ciudad de México 14610, Mexico
Jorge Melendez-Zajgla,Génómica funcional del cáncer, Instituto Nacional de Medicina Genómica, Ciudad de México 14610, Mexico
Jorge Olmos,Biotecnología Marina, Centro de Investigación Científica y de Estudios Superiores de Ensenada, Ensenada 22860, Mexico
Abstract
Key Words: Breast cancer stem cells; Platelets; Metastasis; Tumor microenvironment; Epithelial-mesenchymal transition
INTRODUCTION
Cancer is a multifactorial disease associated with a broad spectrum of molecular alterations inside the cells and the cellular microenvironment[1]. Cancer progresses mainly due to (1) uncontrolled cellular division; (2) alterations in the mechanisms that regulate cell death; (3) epithelial-mesenchymal-transition; (4) intravasation of tumor cells into the circulatory system; (5) cell migration to distant sites; (6) extravasation of tumor cells to initiate new foci; and (7) metastatic tumor development[2].
Breast cancer (BC) is a complex disease characterized by high cellular heterogeneity that comprises[3]a tumor microenvironment[4]and a stem cell niche[3]. BC is the leading cause of cancer death in women worldwide[5]. The lymphatic system is the main system involved in breast tissue drainage[6]. In BC, this is of high relevance due to its role in tumor cell dissemination and subsequent metastatic tumor development. The flow rate through the lymphatic system is 100 to 500 times greater than through blood vessels, and its shear force is lower due to the higher dilation capacity of lymphatic vessels; this means that metastatic cells traveling through the lymphatic system are much more likely to succeed in colonizing a second microenvironment[7].
BC is one of the most studied cancers; however, several molecular processes associated with BC are still unknown. There are various risk factors associated with BC development, such as estrogen production by menopause, or by nulliparity, a sedentary lifestyle, alcoholism, obesity, ionizing radiation, hormonal therapy, age, sex, family history, among others[8,9]. According to a report by The Global Cancer Observatory(a division of the World Health Organization), in 2018, there were 2088849 incident cases of BC and 626679 deaths worldwide[5].
Despite the tremendous histological diversity of breast tumors, a molecular classification system has been developed based on the expression of progesterone receptors (PR), estrogen receptors (ER), and the epidermal growth factor-2 (HER2)[10]. This classification system divides BC tumors into luminal-A, positive for ER and positive for PR, but negative for HER2; Luminal B, ER-positive, PR-negative, and HER2-positive; HER2, ER-negative, PR-negative, but HER2-positive. Triple-negative (TNBC) or basal-like, ER-negative, PR-negative, and HER2-negative[10].
PR and ER are expressed in the membrane of tumor cells and depend on their ligands to proliferate[11]. HER2+ tumor cells have many copies of theHER2gene and high levels of theHER2protein, which probably play a role in the accelerated growth of tumors[12]. Breast tumors classified as triple-negative are common in Black and Latino ethnic groups and are found more frequently in younger women[13].
The treatment and prognosis of a patient are closely related to the molecular subtype of BC present[10]. It has been reported that the subtype with the best prognosis is luminal A, while the one with the worst prognosis is triple-negative[14].
Breast cancer has a highly complex biological and molecular behavior as each cell subtype sends different signals to the tumor microenvironment, in this sense, each signal contributes in a different way to cancer cell differentiation, tumor growth rate and metastasis development. Also, BC stem cells and platelets are two crucial players that participate in BC malignancy and metastasis development, transferring signals and regulation factors through the whole process.
BREAST CANCER STEM CELLS
Breast cancer stem cells (BCSCs), also called breast cancer-initiating cells, are a subpopulation of cells within the tumor that can self-renew and produce different tumor cell lines by asymmetric division. The BCSC population is responsible for developing and maintaining the tumor mass through the expression of survival factors, proliferation and migration[3,10]. BCSCs are regulated through interactions with growth factors and cytokines produced by mesenchymal stem cells (MSCs), cancerassociated fibroblasts (CAFs), tumor-associated macrophages (TAMs) and the extracellular matrix (ECM)[15]. These interactions help stimulate CSC self-renewal, induce angiogenesis and promote invasion by tumor cells and their migration towards new tumor foci.
BCSCs have unique characteristics, including an unlimited capacity for self-renewal, resistance to chemotherapy, the ability to induce the formation of new blood vessels to feed the tumor[3,15]and due to their plasticity, the ability to transition between two states reversibly (epithelial-mesenchymal transition, EMT), which allows them to migrate through the lymphatic and blood systems and establish metastatic foci in distant tissues with the help of platelets[3,16].
Two main models explaining tumor origin have been described: (1) The stochastic or clonal evolution classic model does not contemplate CSCs existence; in this model, each cell can induce tumor development throughout mutations. The model assumes that cancer cell clones produced by genomic abnormalities randomly accumulated could explain heterogeneity[3,17]; and (2) The tumorigenesis model considers CSC existence and postulates that cancer arises from stem cells with unlimited self-renewal capacity. According to this model, cancer originates from poorly differentiated cells with epigenetic mutations and unlimited replication capacity[3,18].
BCSCs can be distinguished from other tumor cells by the presence of cell surface markers EpCAM+, CD24-/Low, and CD44+[16,19]. EpCAM, or epithelial cell adhesion molecule, is a transmembrane glycoprotein that participates in intracellular signaling, proliferation, differentiation, and tumorigenic and metastatic processes[20]. CD44 regulates cell-cell and cell-ECM interactions through hyaluronic acid. It participates in cell adhesion, proliferation, survival, and differentiation[21]. CD44 plays an essential role in cancer development as it is responsible for maintaining the multipotentiality of BCSCs[21]. CD24 is a sialoprotein that participates in adhesion, proliferation, and metastasis; its upregulation inhibits the activity of BCSCs[22].
CD49f and ALDH1 are other cell surface markers found in BCSCs that are associated with chemoresistance; therefore, their presence is associated with a poor prognosis and reduced patient survival. CD49f, also known as α6-integrin, binds to laminin and facilitates the adhesion of epithelial cells to the ECM. ALDH1 is a cytosolic isoenzyme that catalyzes the oxidation of retinol to retinoic acid[23].
Liet al[3]indicated that all mammary tumor subtypes are produced by luminal stem cells. According to these authors, the different mammary tumor subtypes have a single origin that, due to different epigenetic and oncogenic events, are dispersed into the different known BC subtypes.
Different cell surface markers have been observed in BCSCs depending on the cells' EMT state[16,24]. EMT is a process characterized by the loss of apicobasal polarity, loss of intracellular junctions and ECM junctions, and changes in the cytoskeleton[16,25]that make BCSCs, which have epithelial morphology, acquire a mesenchymal phenotype that allows travel through the blood or lymphatic vessels to reach distant sites and create metastatic foci. The phenotype of BCSCs in the EMT state is CD24-CD44+Ecadherin-EpCAM- Vimentin+ALDH+. BCSCs that are in the mesenchymal-epithelial transition (MET) state have a phenotype E-cadherin+EpCAM+and Vimentin-[19,24,26]. The transition between these states is induced by several factors contained in the tumor microenvironment[16,24]. The inflammatory immune response and hypoxia[27], for example, are EMT inducers in BCSCs. Transforming growth factor β (TGF-β) induces EMT by downregulating epithelial proteins such as E-cadherin and upregulating factors like Twist and Snail, which induce the mesenchymal state[28-31]. TGF-β also supports BCSC functionsviaWnt[32]and FAK[33]and promotes the expression of vimentin[26].
Labelleet al[25]reported that BC cells exposed to platelets treatment developed a mesenchymal phenotype; additionally, when these cells were also subjected to treatment with TGFβ, their invasive and metastatic capacity increased. These results indicate that platelets and TGFβ act synergistically and can produce cells with characteristics of BCSCs[25].
In 2014, Asieduet al[34]showed that AXL upregulation by a member of the TAM family (Tyro3, Axl, Mer; tyrosine kinase receptors) increases the tumorigenic capacity, invasion, and metastasis of BC cells, while AXL downregulation reverses EMT in BCSCs and restores chemosensitivity. AXL regulates several signal transduction pathways, including NF-k β, STAT, Akt, and MAPK. Asideuet al[34]demonstrated that AXL is constitutively activated in BCSCs and that it induces EMT by regulating Ecadherin, N-cadherin, Snail, and Slug expression.
It has been shown that BCSC activity is also regulated by signals from the Notch, Hedgehog, Wnt/ β-catenin, p53, and TGF-β pathways[16,32], helping to maintain cell survival and proliferation and, in this way, contributing to the growth of breast tumors.
TUMOR MICROENVIRONMENT
The tumor microenvironment (TME) is constituted by the ECM, CAFs, MSC, TAMs, and inflammatory cells (ICs)[35]. The TME is also constituted by soluble active biomolecules such as cytokines, growth factors, ligands, membrane-anchored molecules, secretion proteins and RNAs (miRNAs, lncRNAs)[28,36](Figure 1).
All the interactions that dictate the behavior of the tumor take place in the TME. This behavior can include a reversion to healthy tissue states or progression to the most advanced and deadly stages of the disease. Some of the interactions in the TME that promote cancer development, such as angiogenesis and metastasis, are discussed below.
The ECM provides attachment sites to normal and tumor stem cells, allowing them to interact with signals from other cells or from the TME that plays a role in their maintenance and regulation. It is through the ECM that CSCs that have experienced EMT can exit to points distant from the primary tumor[24]. It has been reported that in BC, the ECM increases its rigidity, which promotes TAZ protein activation[36], one of the main effectors of the Hippo pathway, responsible for controlling organ size by regulating cell proliferation, apoptosis and stem cell renewal[37].
MSCs regulate BCSCs through the loops of cytokines IL-6 and CXCL7; IL-6 is produced by MSCs and other immune cells present in the TME[38]. The interaction between IL-6 and the ILR 6/gp130 receptor present in BC cells induces CXCL7 production. In turn, CXCL7 induces IL-6, IL-8, CXCL6 and CXCL5 secretion, all of them with the ability to stimulate the self-renewal of BCSCs, which leads to tumor growth, metastasis and chemotherapy resistance[39].
TAMs are also part of the TME; they accumulate in hypoxic microenvironments and are the main orchestrators of an inflammatory TME. They promote cancer cell proliferation, invasion, and metastasis, stimulating angiogenesis and inhibiting the Tcell-mediated antitumor immune response. Furthermore, they play a crucial role in the regulation of EMT[40].
TAMs secrete TNF-alpha[41], which induces EMT through the NF-κβ pathway[42], and also induce an increased expression of Slug, Snail, and Twist[40]. Furthermore, TAMs indirectly increase the self-renewal capacity of BCSCs by maintaining constant communication with TGF-β[40].
To support tumor angiogenesis, TAMs produce angiogenesis-modulating enzymes[43]such as the metalloproteinases MMP-2, MMP-7, MMP-9, MMP12, and cyclooxygenase 2[43,44]. They also release cytokines (CXCL12, CCL2, CXCL8, CXCL1, CXCL3, and CCL5) that play a crucial role in different processes associated with cancer development[45].
Micro-RNAs (miRNAs) are also associated with the TME. These molecules are small non-coding RNAs that participate in various cellular functions, are found in large numbers in the TME, and are essential for the maintenance, development, and progression of cancer[36]. They are also critical regulators of the signaling pathways associated with cell stemness. The presence of oncogenic miRNAs in the TME is partly explained by their increase during specific transcription and by their release by tumoreducated platelets[46,47].
Several miRNA clusters, such as miR-183, miR-221–222, let-7, miR-142, and miR-214, play a role in the maintenance of BCSCs[47]. Others, such as let-7, miR-7, miR-10, and miR-15a, have been associated with BCSC chemoresistance ability[48]. EMT and MET transitions are also regulated by miRNAs clusters, including mir-9, mir-100, mir-221, and mir-155 as EMT inducers, while mir-200, mir-205, and mir-93 induce MET[49]. MiR-939 induces the EMT process by reducing E-cadherin and Claudin expression[50].
The p53 protein depletion in tumor cells reduces miR-200c expression[51], induces EMT development, and provides stem cell-like properties. The p53 activation reduces the expression of Snail and other EMT-inducing transcription factors through the upregulation of the miR-34 family[52].
The miRNAs found in the TME can come from platelets or platelet microparticles (PMPs). PMPs are the most abundant microparticles in the blood and contain, in addition to miRNAs[53], several EMT-promoting factors, including basic fibroblast growth factor (bFGF), fibroblast growth factor (FGF)[54], platelet-derived growth factor (PDGF)[55], hepatocyte growth factor (HGF)[56], TGF-β[57], vascular endothelial growth factor (VEGF)[58]and brain-derived neurotrophic factor/tropomyosin-related kinase (BDNF/TRK)[59].
TGF-β secreted by PMPs allows the overexpression of miR-183, which decreases the expression of DAP12, a receptor of natural killer cells (NKs) that plays a crucial role in membrane potential stabilization and signal transduction in these cells. The result is an increase in the survival of migratory cancer cells within blood vessels[60].
The transcription factor NF-kB participates in the regulation of metastasis and the induction of EMT. The activation of NF-κB is associated with a change of the TGFβ factor from a tumor-inhibiting role to a prometastatic role. The direct contact of platelet-derived TGFβ with tumor cells activates the TGFβ/Smad and NF-κB pathways in cancer cells, promoting EMT, and increasing metastasisin vivo[25,57].
It has been widely reported that a chronic inflammatory microenvironment promotes tumor development[4]. These inflammatory microenvironments are usually enriched with several lipid mediators such as platelet-activating factor (PAF)[61], prostaglandin (PG)[62]and lysophosphatidic acid (LPA)[63], all of which are secreted by platelets, by different types of immune cells during inflammation and by BC cells after stimulation with growth factors. These lipid mediators play an essential role in platelet aggregation and neo-angiogenesis[61-63]. The PAF has been shown to play an essential role in the onset and progression of BC and plays a predominant role in neoangiogenesis[61]. Wardet al[64]reported that LPA release by the GPCR CD97 complex activates platelets and improves the permeability of tumor cells within blood vessels, which results in a higher capacity for invasiveness[64].
ROLE OF PLATELETS IN CANCER PROGRESSION AND METASTASIS
Platelets were discovered by the Italian physician Giulio Bizzozero in 1882. In 1906, JH Wright described them as anuclear cell fragments derived from megakaryocytes[65].
Platelets are widely known as the most critical first response factor in (1) coagulation, vasoconstriction, and inflammation, facilitating sterilization, tissue repair, and resolution; (2) they are the first to detect, phagocytize and react to pathogens in the circulation; (3) they maintain vascular integrity by hemostasis; (4) heal wounds; and (5) initiate and coordinate intravascular immune responses during infections and cancer[66,67].
The secretome of platelets is made up of more than 300 biomolecules, mainly in dense granules, alpha granules, and lysosomes. The presence of these biomolecules in the granules may be explained by the endocytosis and biosynthesis processes that take place in the megakaryocyte from which platelets originate[68].
One of the properties of platelets as first responder cells is the ability to actively migrate through any inflamed or leaking vessel wall, in response to a variety of stimuli, in order to aid in wound sterilization and tissue regeneration[66](Figure 2).
Platelet morphology changes according to the state they are in. When they are in a resting state, platelets assume a discoid shape that maximizes interactions with flat surfaces[69,70]. In this state, platelets travel out of blood vessels and, after coming into contact with external microenvironmental factors, become activated, and their cytoskeleton undergoes a series of changes that result in the development of filopodia, which enlarge the cell contact surface and allows platelets to provide a highly rapid response to tissue injuries[70].
There are two ways by which platelets manage to disseminate the information they collect from the various microenvironments in which they are present. The first is by shedding membrane-enclosed cell fragments or microvesicles such as PMPs. These are also known as platelet-derived microvesicles (PMVs), which range in size from 100 to 1000 nm or others such as exosome-like microvesicular bodies range in size from 40–100 nm, that carry a wide variety of biomolecules such as miRNAs, growth factors and cytokines, among others. The second way by which platelets disseminate information is membrane fusion[71].
The platelet content is enriched by the various microenvironments through which they circulate. When circulating through the TME, platelets incorporate tumorassociated molecules, including molecules from TMAs or CSC niches that can support the growth of the original tumor or help establish secondary metastatic foci. These platelets are called "tumor-educated platelets" (TEPs)[72].
The molecules secreted by platelets include nitric oxide (NO), a bioactive compound that modulates angiogenesis, the immune response and neural regulation[73]. It is synthesized by nitric oxide synthase (NOS2) during the oxidation of L-arginine and has been shown to decrease the apoptosis of human lung carcinoma cells. In BC, NOS2 is a biomarker of disease progression and prognosis. NOS2 also mediates angiogenesis and the immune response in the TME, which are critical factors in cancer development[73,74].
In 2016, Banskotaet al[75]demonstrated that serotonin is also released by platelets, inducing the production of reactive oxygen species (ROS) derived from NADPH oxidase (NOX) in tumor cells[75]. ROS production occurs as a result of alterations in the functions of mitochondria, associated with the development and progression of various diseases, including cancer[76]. In cancer, ROS production promotes the activation of the epidermal growth factor receptor (EGFR), which has been identified as a tumorigenesis driver and tumor resistance biomarker[77]. EGFR is capable of inhibiting anoikis, a form of programmed cell death caused by the detachment of cells from the ECM through activation of the extracellular signal-related kinases (ERK) pathway[78], which is associated with cell proliferation, migration, apoptosis, differentiation and senescence.
Along with ATP[67,79], platelets release other metabolites, such as thromboxane A2 (TxA2), 12-HETE[80]and serotonin[65], which, by regulating the permeability of blood vessels, allow TEPs and other cells with a migratory phenotype to move through blood vessels and colonize secondary metastatic foci[72,79]. In 2013, Schumacheret al[81]demonstrated that TEPs release adenine nucleotides (ATP) from their dense granules, which act on the P2Y2 receptors of endothelial cells, disrupting the adhesive junctions of endothelial cells and opening the endothelial barrier of blood vessels, which allows tumor cells to exit the bloodstream and travel to new metastatic foci[81].
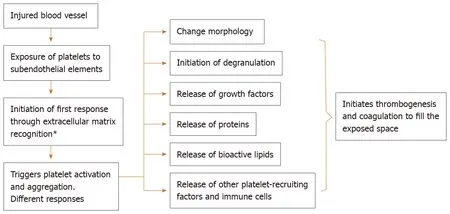
Figure 2 The role of platelets in wound biology.
The platelet's role in coagulation has been widely studied. Tissue Factor (TF), also called platelet tissue factor, is a transmembrane glycoprotein that binds to the coagulation serine protease factor VII/VIIa (FVII/VIIa) and is activated when a blood vessel is injured, triggering the coagulation cascade (extrinsic pathway). Fibroblasts typically express TF on blood vessel walls[82]. Its presence promotes thrombin production and platelet activation, which has been associated with cancer, contributing to the survival of cancer cells and metastasis[83]. In 2015, Orellanaet al[84]showed that exposure to platelets increases TF expression in cancer cells[83]. As cancer cells can also express TF on their cell membrane, they can activate the coagulation cascade, which leads to the production of thrombin and the activation of platelets[83,84]. Thrombin is a crucial mediator of the metastatic process participating in both angiogenesis and cell migration and is capable of inhibiting apoptosis and inducing the proliferation and differentiation of vascular progenitor cells[85].
It has been reported that endothelial cells stimulated by thrombin induce morphology changes related to an EMT phenotype, which, as discussed, is associated with the loss of adhesive cell-cell junctions and migration to blood vessels[86].
Platelets also express multiple receptors on their surface that are activated according to signals they receive from the microenvironment. These receptors can generate different responses, including hemostasis, thrombosis, inflammation and tissue remodeling, but also the promotion of cancer cell survival and metastasis[67,72]. Mice that have been depleted of platelets or have a deficiency in granule content do not develop metastasis[87]. Other studies have shown that the proteins released by platelets influence the degree of malignancy of breast tumor cells, contributing to the generation of a more aggressive and metastatic phenotype[88].
Once CSCs acquire the migratory phenotype through EMT and are ready to start moving to sites distant from the primary tumor, they must be protected by platelets from shear forces and the first line of defense of the immune system in blood, NK cells[89,90](Figure 3). Migratory cells activate platelets through different mechanisms, in an exacerbated manner, which explains hypercoagulation and the increased risk of thrombosis in cancer patients[66,68].
The chemokines and cytokines that stimulate cell migration, CXCL1, CXCL4, CXCL5, CXCL7, CXCL8 and CXCL12, play an essential role in the platelet response to tissue injury, but also play a key role in angiogenesis and the metastatic process of BC[91]. CXCL4, also known as platelet factor 4 (PF4), is produced by megakaryocytes and stored in platelet α granules during platelet formation. The primary function of this chemokine is to contribute to coagulation but, being an angiostatic factor, it also plays a decisive role in the development of cancer by inhibiting cell migration[91,92].
Johnsonet al[93]looked for a relationship between the biomolecules secreted by platelets and some type of secretome in BC cells that could make them prometastatic. They found that platelet-secreted biomolecules in the TME induced the release of CCL2, angiogenin, interferon-gamma, IL-6, granulocyte-macrophage colonystimulating factor and CXCL1 in BC cells. They also found that IL-8 or CXCL8, the most studied proinflammatory chemokine secreted by metastasis-promoting tumor cells, increases its expression 50-fold in BC cell lines[93]. This cytokine is known to be released in response to platelet activation and aggregation[43]. IL-8 interacts with the CXCR1 and CXCR2 membrane receptors, which are highly expressed in BC cells; in fact, their expression is associated with angiogenic and metastatic processes, as well as with the regulation of BCSC expansion. Blocking CXCR1 and CXCR2 receptors have an antitumor effect[94].
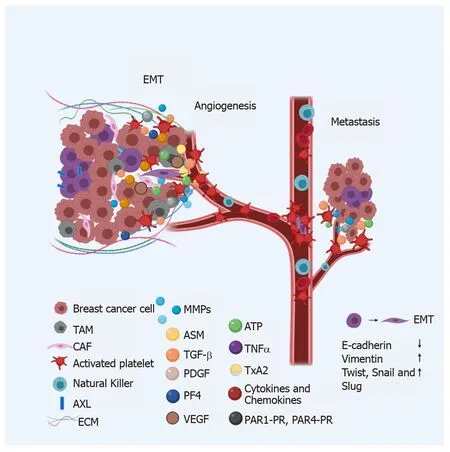
Figure 3 Breast cancer metastasis.
Platelet inactivation in the TME has been shown to inhibit the Akt pathway, which usually induces IL8 production. When the production of IL-8 decreases, the metastatic capacity of BC cells decreases[67]. IL-8 positively regulates the AKT pathway, generating a much more metastatic and aggressive BC phenotype[67]. Consecutively, AKT induces SOX2 overexpression in CSCs, one of the main transcription factors involved in stem cell self-renewal[95].
It has been reported that aspirin can induce platelet inhibition[64,67]. A study by Johnsonet al[93]showed that administering aspirin to BC patients significantly decreased IL8 levels and the platelet count. Aspirin intake was also associated with a decrease in tumor invasion compared to patients who did not receive aspirin[67]. Rothwellet al[96]concluded that people who consume aspirin daily are less likely to be diagnosed with cancer and show a higher probability of survival if they develop malignancy[96].
CCL5, also known as RANTES, is another chemokine associated with the progression and metastasis of BC promoted by platelets. Several authors have shown that platelets store and release large amounts of this chemokine, which is associated with disease progression and metastatic processes in patients with BC[44].
CXCR2 and its ligands are also involved in cancer development by promoting tumor angiogenesis and chemoresistance[95]. It has been reported that the expression of CXCR2 ligands increases in the supernatants of BC cells treated with chemotherapy and that BCSCs are enriched during doxorubicin and paclitaxel chemotherapy, showing more significant metastatic potential than primary tumor cells[97].
Cells from metastatic BC are rich in CXCR4 and CCR7 receptors and it has been observed that the sites where secondary metastatic foci are more likely to develop are those sites rich in ligands for CXCR4 and CCR7[98].
CXCR4 also interacts with CXCL12, a chemokine produced by stromal cells whose leading role is to trigger platelet migration into extravascular spaces, which promotes the polymerization of actin in the cytoskeleton of cancer cells, thereby causing a rearrangement of the pseudopodia of BC cells, which in turn induces chemotactic and invasive responses[99].
PAR1-PR and PAR4-PR (molecules found inside platelets) are other plateletsecreted molecules that play a crucial role in the TME and the development of cancer, angiogenesis and metastasis[66,78]. Their expression increases BC cell proliferationin vitroand, together,in vivo,promote MDA-MB-231 growth implanted in mice[66]. PAR1-PR is a proangiogenic factor that stimulates platelet activation and induces the release of SDF-1 and VEGF by promoting the growth of BC cells, more efficiently[78]than PAR4-PR, an antiangiogenic factor that promotes the release of PF4 and endostatin but limits the release of SDF-1 and VEGF[78].
VEGF, another factor released by platelets[100], promotes cancer cell proliferation mainly through the cooperative signaling of VEGFR2 and integrin activation by VEGF,viathe PI3K/PKC signaling pathway[101]. Breast cancer patients submitted to several treatments showed platelet phenotype changes. Platelets were activated by ADP receptors, thrombin and collagen from patients with BC, which led to a significant increase in the secretion of VEGF, TSP1 and TGF-β1, compared to resting platelets. The response, however, differed depending on the platelet agonist. For example, the ADP agonist was a much weaker inducer of protein release than agonists attacking collagen and thrombin receptors. The study demonstrated the effects of specific stimulation of platelet receptor pathways on the release of VEGF, TSP1 and TGF-β1 in BC patients[102].
In 2015, Orellanaet al[83]studied the association between platelets and ovarian cancer stem cells. They showed that platelets promoted the formation of ovarian cancer spheres that express cancer stem cell surface markers, such as CD44. In addition, the authors showed, through an experiment with a Boyden camera, where cancer cells were placed on top and platelets on the bottom, that microparticles from platelets and exosomes in the proximity of tumor cells promoted the migration of these cells, probably because the molecules inside platelets, such as PDGF and TGFβ, can serve as chemoattractants[83].
Platelets have been reported to induce activation by dephosphorylation and subsequent entry into cell nuclei, of RhoA-(myosin phosphatase targeting subunit 1), MYPT1-protein phosphatase (PP1)-mediated and the Yes-associated protein 1 (YAP1), which confer resistance to anoikis[103]. It has also been shown that platelets with a low expression level of apoptosis signal-regulating kinase 1 (Ask1), which activates the phosphorylation of AKT, JNK and p38, are associated with a decrease in metastasis[104].
Another protein secreted by platelets that plays an essential role in the metastatic process of BC is acid sphingomyelinase (Asm), which induces ceramide production, which activates integrin 51, which in turn promotes metastasis[105]. Ferroniet al[106]demonstrated that cancer-associated oxidative stress contributes to persistent platelet activation.
In TNBC, the most aggressive form of BC with the poorest outcome, there are molecules related to self-renewal signaling pathways that are highly activated in TNBC relative to non-TNBC, these include SRC, PTK7, CX26, USP2, and PLK1[107].
On the other hand, Janssonet al[108]reported that the platelet-derived growth factor (PDGF) has higher expression in the TNBC subtype and the prognosis of these patients is even worse. Also Camoraniet al[109]considered PDGFRß as "a reliable biomarker of TNBCs subgroup with invasive and stem-like phenotype[109]." In this sense, several PDGF receptor kinase inhibitors have been developed, including Imatinib, Sunitinib, Sorafenib, Pazopanib, and Nilotinib. In addition, monoclonal antibodies directed against PDGF or PDGFR have been developed, such as MC-3G3 specific to PDGFRα, and IMC-2C5 directed against PDGFRβ to delay tumoral growth[110].
Experimental evidence indicates that platelets can exacerbate the cancer metastasis process in several ways; (1) promoting extravasation; (2) improving tumor cell survival in the circulatory system; (3), increasing tumor cell arrest in the vasculature system; and (4) stimulating tumor proliferation and angiogenesis at secondary sites[111].
ROLE OF PLATELETS IN CANCER CELL PROTECTION
When cells metastasize, only a small group of them manage to survive and initiate metastatic foci. Platelets play an essential role in cancer cell survival, by protecting anoikis. When cancer cells travel throughout the bloodstream, platelets induce thrombus formation to protect them from dangerous shear forces and NK cells[71,91,111,112]. Anoikis is an apoptotic process induced by cell detachment from the ECM that constitutes the main barrier against the metastatic process[112]. As discussed above, one of the main characteristics of cells with a migratory EMT phenotype is the loss of cell-cell and cell-ECM junctions, which allows them to enter the blood or lymphatic vessels and colonize distant sites as part of the metastatic process[16,24]. A characteristic of cancer cells that has been fully identified is their ability to survive without anchorage, travel to distant sites through the blood system and initiate secondary tumors[16]. Several studies have analyzed the role of signaling pathways in anoikis. For example, PI3K-AKT is one of the main pathways involved in promoting resistance to anoikisviathe growth factor IGF-1, which is found in the TME and plays an essential role in tumor development and growth, as well as in the prevention of apoptosis through its ligand IGFR1[113].
In 2002, Zenget al[77]showed that the cytokine HGF, which promotes cell proliferation, migration and invasion, can inhibit anoikis and improve the survival of head and neck tumor cells through activation of the ERK pathway[77].
In 2004, Doumaet al[114]demonstrated the role of the neurotrophic tyrosine kinase receptor TrkB as a direct suppressor of caspases in anoikis. TrkB prevents cell death, allowing them to survive and proliferate as suspended spheroid cell aggregates[114].
A large number of adhesion molecules embedded in the cell membrane of platelets and platelet-derived microparticles promote adhesion between different cells; platelet/platelet, platelet/blood vessel endothelium and platelet/cancer cell. The cellular adhesion aids the formation of a thrombus around cancer cells that have entered blood vessels and thereby helping them evade both anoikis and the immune response[115]. The adhesion molecules of platelets include integrins (αIIbβ3, α2β1, α5β1, α6β1, αLβ2, αvβ3, P-selectin), membrane glycoproteins (GPIb/V/IX and PSGL-1) and immunoglobulin superfamily proteins (platelet-endothelial cell adhesion molecule-1, PECAM-1)[115,116].
Both platelets and endothelial cells have been shown to express P-selectin, which aids the dissemination of tumor cells. P-selectin can be found in the alpha granules of platelets. When platelets are activated, P-selectin is translocated to the surface and regulates the binding between platelets and endothelial cells through the formation of glucan structures[117]. It has been shown that a decrease in P-selectin, which weakens the junctions between platelets and tumor cells, is associated with a reduction in tumor size, a reduction of metastatic foci and an increased survival rate in mice with BC[118]. P-selectin is known to play an essential role in the evasion of NK cells by BCSCs in blood vessels. Platelets aggregate around a tumor cell when the PSGL-1 ligands on platelets recognize P-selectin, allowing tumor cells to evade the immune response[119].
ROLE OF PLATELETS IN ANGIOGENESIS DEVELOPMENT
As neo-angiogenesis produces leaky blood vessels during the early progression of cancer, it is no surprise that platelets are among the first cells to be involved in this process. Their ability to extravasate, activate and release proangiogenic, chemoattractant and immunomodulatory compounds can potentially promote cancer progression and metastasis.
Neo-angiogenesis is regulated by several pro- and antiangiogenic factors, mainly VEGF[120], FGF[103], EGF[121], HIF-1[122]and TGF[123], but many other angiogenic cytokines such as factors PDGF[103,120], NGF[124]and SCF[125]participate in this process.
In cancer, the angiogenesis process is promoted and regulated by the TME, including BCSCs. Two mechanisms explain the formation of new blood vessels in tumors. The first involves the transdifferentiation of cancer cells in a process called vasculogenic mimicry[126]. The second one involves the binding of neoplastic cells to the vessel wall through a process called mosaic vessel formation[127]. Both mechanisms depend on stimulating factors that promote the formation of new vessels. Fenget al[128]reported that the vesicle-associated membrane protein 8 (VAMP8), which can be found in platelet α-granules, is capable of attracting and recruiting bone marrow cells to hypoxic stress points in the tumor tissue, thus contributing to the formation of blood vessels within the tumor[128].
PLATELETS AND STEM CELLS INTERACTION
In mice, platelets help stem cells promote the proliferation of cells in injured tissues. They also help differentiate them through platelet-derived factors such as SDF-1, which not only regulates stem cell adhesion but also promotes the differentiation of CD34+ cells into the EPC under strong shear forces[129]. Healthy stem cells travel through the bloodstream, leaving it to reach injured tissue and initiating replication, differentiation and repair. Cancer stem cells, originating in the core of the tumor and having undergone a process of cell detachment and MET, allow them to invade distant tissues and go through the same type of processes as healthy stem cells. In both cases, platelets help stem cells survive and reach their target site, in one case, to repair injured tissue and in the other to colonize and form metastatic foci. Healthy stem cells have not been reported to activate the platelets that protect them, but several studies have shown that cancer stem cells continuously activate platelets through various pathways.
Interestingly, bone marrow-derived mesenchymal stem cells (BM-MSCs) produce cytokines and exosomes, promoting tumor growth and metastasis of cancer cells. Recently, researchers reported that BM-MSCs presented transdifferentiation TGFß dependent into CAFs and perivascular-like cells after co-incubation with platelets which was associated with an overexpression of vimentin, fibroblast activation protein and a-smooth muscle actin. Transdifferentiated-BM-MSCs cell medium had an interesting effect on gastric cancer cells: they were able to metastasize to lung and increased their proliferation and migration toward cancer cells[130-132].
Recently, efforts to create new therapies have been focused on targets related to pathways involved in stemness of BC cells, many of which have already been reviewed in this work. These factors can be transported into platelets or they can be regulated by the platelet content released into the microenvironment. Some of the developed therapies are shown in Table 1.
On the other hand, several clinical studies have suggested that treatment with anticoagulants (AC) or antiplatelets (AP) helps treat cancer by directly influencing platelet behavior and indirectly affecting tumor cell behavior. A large cohort study of patients showed that daily use of aspirin, the most commonly used antiplatelet agent was associated with a reduced incidence of malignancy[133], and this effect was evident in colorectal, prostate and breast cancer[134,135]and for others types of cancer there is still controversy.
Other antiplatelets in addition to aspirin have been studied, such as Dipyridamole and RA-233 in pancreatic cancer; Prasugrel in gastrointestinal cancer, Clopidogrel in the pancreatic cancer mouse model and hepatoma carcinoma, melanoma and breast cancer with promising results. It is clear that more studies are needed in order that personalized platelet-targeted therapies in cancer may be administered.
CONCLUSION
TEPs can be considered the perfect weapon of CSCs for tumor development and metastatic foci formation.
TEPs contribute to various levels of cancer progression when platelets have been activated by stimuli from TME or cancer cells, they secrete factors that strengthen the TME by promoting (1) the mobilization of tumor cells into blood vessels, to produce metastatic foci; (2) tumor growth; and (3) neo-angiogenesis to feed the tumor.
In order to be able to contribute to cancer mortality reduction, it is necessary to understand the interactions between platelets and BCSCs in the TME, and how they control cancer progression and metastatic secondary tumor development. This information could be useful in identifying new therapeutic targets or in the development of an accurate and straightforward diagnosis.
杂志排行

World Journal of Stem Cells的其它文章
- Acquired aplastic anemia: Is bystander insult to autologous hematopoiesis driven by immune surveillance against malignant cells?
- Glutathione metabolism is essential for self-renewal and chemoresistance of pancreatic cancer stem cells
- Effect of conditioned medium from neural stem cells on glioma progression and its protein expression profile analysis
- Immunophenotypic characteristics of multipotent mesenchymal stromal cells that affect the efficacy of their use in the prevention of acute graft vs host disease
- AlCl3 exposure regulates neuronal development by modulating DNA modification
- Isolation and characterization of mesenchymal stem cells in orthopaedics and the emergence of compact bone mesenchymal stem cells as a promising surgical adjunct